



Hereditary spastic paraplegia is a group of inherited neurological disorders with predominant manifestations of lower extremity weakness and severe spasticity. This is a genetically heterogeneous disorder very difficult to distinguish clinically with many genes described. Few patients with this condition have been previously reported.
Patient and methodsWe present a case of a 5 years old girl, born from consanguineous parents, with severe ataxia and progressive spasticity of low limbs. Due to the severity of the symptoms and the need for early diagnosis, next generation sequencing study of 37 genes implicated in spastic paraplegia was performed.
ResultsA novel pathological variant in FA2H gene was discovered. Father, mother and brother were heterozygous carriers.
ConclusionsSpastic paraplegia due to mutations in FA2H is an under diagnosed condition, and it should always be considered in childhood onset of progressive pyramidal dysfunction. Next Generation Sequencing allows a simultaneous analysis of many genes, enables a fast diagnosis in complex disorders.
La paraparesia espástica es un grupo de enfermedades neurológicas hereditarias que cursan con debilidad de las extremidades inferiores y espasticidad severa. Es una enfermedad muy heterogénea, con muchos genes descritos y muy difícil de distinguir clínicamente. Hay pocos pacientes descritos con esta enfermedad.
Pacientes y métodosSe presenta un caso de una niña de 5 años, de padres consanguíneos, con una ataxia severa y espasticidad progresiva de los miembros inferiores. Dada la gravedad de la clínica y la necesidad de un diagnóstico temprano, se decide realizar un panel de secuenciación masiva de 37 genes implicados en paraparesia espástica.
ResultadosLos resultados muestran una variante patológica no descrita previamente en el gen FA2H. El padre, la madre y el hermano resultan portadores heterocigotos.
ConclusionesLa paraparesia espástica debida a mutaciones en el gen FA2H está infradiagnosticada y debería ser considerada siempre que aparezcan síntomas en la infancia de disfunción piramidal grave y progresiva. Los paneles de secuenciación masiva con el análisis simultáneo de varios genes están permitiendo un diagnóstico más rápido en enfermedades complejas.
Hereditary spastic paraplegias (HSP) are a large group of clinically heterogeneous neurodegenerative disorders characterized by severe spasticity and weakness of lower limbs. The prevalence of HSP is estimated in a range from 1.3 to 9.6 per 100,000. The condition is defined as pure or complicated if there is a presence of other neurological features. The most useful classifications now are based on the mode of inheritance and genetic linkage. Genetic defects have been identified in more than 40 different genes with X-linked, recessive, dominant and maternal inheritance.1
To date, the locations of several genes associated with HSP have been identified. The most common cause of autosomal dominant spastic paraplegia are SPAST/SPG4 mutations, with patients presenting with a pure form of HSP.1 In the autosomal recessive complex HSP, the most frequent form is associated with thinning of the corpus callosum due to mutations in SPG11.2 Recently a complicated form of autosomal recessive spastic paraplegia called SPG35 with early onset was identified due to mutation in the FA2H gene,3 speech involvement, intellectual disability, epilepsy and microcephaly were commonly associated features; symptoms gradually get worse over time.
We report a 5 year-old-girl that presented severe progressive spasticity of low limbs due to a spastic paraplegia Type 35. This is a severe recessive disorder characterized by childhood onset, with a progressive spasticity of lower limbs, mild phenotype, leucodistrophy and brain iron accumulation caused by mutations in FA2H gene.4 To date few patients with FA2H mutations are described. FA2H is involved in the synthesis of 2-hydroxy fatty acid galactolipids, major components of the myelin sheath. It has been demonstrated that abnormal hydroxylation of myelin galactocerebroside lipid components can lead to a severe progressive impairment.5 This is a genetically heterogeneous disorder that is difficult to distinguish clinically. The overlap with other neurodegenerative diseases and the high number of implicated genes make difficult a correct diagnosis in many cases, especially in children. An early diagnosis is required in childhood severe cases not only for treating disease but also for genetic counselling in the family. Next generation sequencing technique (NGS) is transforming and improving the diagnostic approach for neurological disorders, since it allows simultaneous analysis of hundreds of genes with a fast and relative low cost-efficient diagnosis.
Patient and methodsThe patient was a Caucasian 5-year-old girl born from consanguineous parents, they were first cousin, without any familiar history of neurological disorders. They had an asymptomatic second son (Fig. 1). The girl had onset of ataxia and tip–toe waking at 4 years and 9 months of age, few months later she presented low limb spasticity, frequent falls and rapidly progressive degeneration so that she had loss independent ambulation. Her early milestones were reported as normal, metabolic test, echocardiogram and ophthalmologic examination were normal. She had a poor language development, mild cognitive decline without any dysmorphic facial features. MR showed vermian hypoplasia but no iron accumulation or leucodistrophy. Peripheral blood was extracted from patient and parents under informed consent.

DNA from girl, parents and brother was extracted by conventional methods.
Molecular testing was initiated in the girl first for 18 genes involved in hereditary Ataxia with negative results (Hiseq platform Illumina) KCNA1, CACNA1A, CACNB4, SLC1A3, VAMP1, ATM,TTPA, APTX,SETX, C10orf2, SIL1, FTL, POLG, SACS,TDP1, CYP27A1, ABCB7, PRICKLE1. Thereafter, a study for the 37 more frequent spastic paraplegias gene by NGS panel was performed (Hiseq platform Illumina): AP4B1, AP4E1, AP4M1, AP4SI, AP5ZI, ATL1, BSCL2, C12orf65, CYP2U1, CYP7B1, DDHD1, DDHD2, ERLIN2, FAH2H, GBA2, GJC2, K1AA0196, KIF1A, KIF5A, LICAM, NIPA1, PLP1, PNPLA6, REEP1, RTN2, SLC16A2, SPAST, SPG11, SPG20, SPG21, SPG7, TEC, PR2, VPS37A, ZFYVE26, ZFYVE27, HSPD1.
A novel variant was discovered in FA2H gene, c.379C>T (p.Arg127Ter) in homozygosis. This nonsense mutation leads to a stop codon therefore it most probably affects to protein function. Nonsense mutation is a known mechanism of this disease. Segregation of the variant from the parents was confirmed by Sanger sequencing (Fig. 1). The variant has not been previously reported in the single nucleotide polymorphism database. Therefore, this variant was classified as pathogenic according to ACMG guideline.6
DiscussionComplex recessive spastic paraplegias have been frequently associated with mutations in SPG11 (spatacsin), ZFYVE26/SPG15 and SPG7 (paraplegin), however about half of cases remain without a genetic diagnosis indicating the existence of yet unidentified genes.7,8 Mutations in FA2H is recently related with severe neurodegeneration including spastic paraplegia.9 In FA2H-deficient mice studies, those lacking hydroxylated lipids in the central and peripheral nervous system showed late onset axon and myelin sheet degeneration with normal neuronal development.10 It has been also hypothesized that rare deleterious heterozygous mutations of FA2H might constitute risk factors for autism.11
In this report, we described a 5-years-old girl with a rapid deterioration of motor system, she presented with ataxia first, but she could not walk few months later with severe spasticity and weakness of the lower limbs. She had not leucodistrophy neither iron accumulation in the brain. It is unknown what factors could influence the development of iron accumulation in the brain but this feature might appear later as it is described in literature.4 Another possibility is that iron accumulation is caused by apoptotic from cellular damage demyelinating. Iron is crucial for myelination, but excessive amounts of iron could directly contribute to tissue damage. It has been demonstrated in vitro that free iron or iron overload can lead to free radical formation, lipid peroxidation, and neuronal damage.12,13 Iron deposition in patients affected by FA2H mutations appears to be variable, but may become more evident over time.
Both parents were heterozygous carriers of the mutation as well as the bother of the girl. The father was 26 years old and the mother 22. Genetic counselling is essential in this family and relatives for future gestations in which prenatal diagnosis or preimplantational genetic diagnosis can be offered.
Childhood spastic paraplegias are very difficult to diagnose because are a large group of clinically heterogeneous disorders, but especially those recessive because of lack of family history. Specific molecular genetic tests can help in early diagnosis and help patients to avoid invasive and costly clinical testing. Consecutive screening of main candidate genes is normally performed as strategies to diagnose these complex cases but it is costly and time consuming. NGS is faster and permits a cost-effective diagnostic test.14,15 The development of technology is improving the management and prognosis of many diseases due to the short turnaround time. NGS technology permits a simultaneous analysis of hundreds of genes, enable a fast diagnosis even in less informative cases.16
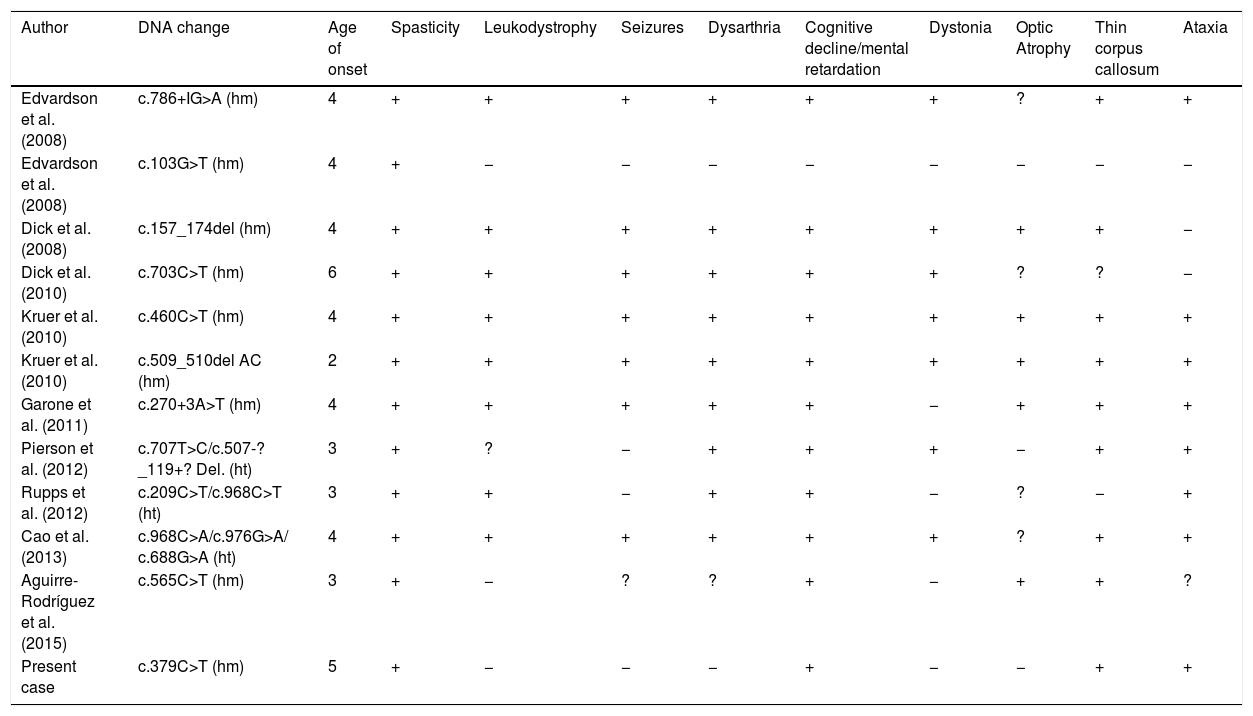
Although some author proposed a correlation of mutation and disease severity5,16 other studies do not correlate the symptoms with the type of mutation.17 Nonsense mutation cause the absence of the protein product leading to severe symptoms, a nonsense mutation was published in a girl with mildly decreased muscle tone in upper limbs and psychomotor development,18 our patient presents low limb spasticity and speech delay. A comparison of the phenotypic features in some previously described patients is showed in Table 1.19–22 Spasticity, thin corpus callosum and intellectual impairment is present in almost all patients. Other features such as dystonia, ataxia, dysarthria and seizures were present in many cases, but the severity of symptoms varied among reported families.
Comparison of the phenotypic features in previously described families with pathogenic variants in FA2H.
| Author | DNA change | Age of onset | Spasticity | Leukodystrophy | Seizures | Dysarthria | Cognitive decline/mental retardation | Dystonia | Optic Atrophy | Thin corpus callosum | Ataxia |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Edvardson et al. (2008) | c.786+IG>A (hm) | 4 | + | + | + | + | + | + | ? | + | + |
| Edvardson et al. (2008) | c.103G>T (hm) | 4 | + | − | − | − | − | − | − | − | − |
| Dick et al. (2008) | c.157_174del (hm) | 4 | + | + | + | + | + | + | + | + | − |
| Dick et al. (2010) | c.703C>T (hm) | 6 | + | + | + | + | + | + | ? | ? | − |
| Kruer et al. (2010) | c.460C>T (hm) | 4 | + | + | + | + | + | + | + | + | + |
| Kruer et al. (2010) | c.509_510del AC (hm) | 2 | + | + | + | + | + | + | + | + | + |
| Garone et al. (2011) | c.270+3A>T (hm) | 4 | + | + | + | + | + | − | + | + | + |
| Pierson et al. (2012) | c.707T>C/c.507-?_119+? Del. (ht) | 3 | + | ? | − | + | + | + | − | + | + |
| Rupps et al. (2012) | c.209C>T/c.968C>T (ht) | 3 | + | + | − | + | + | − | ? | − | + |
| Cao et al. (2013) | c.968C>A/c.976G>A/ c.688G>A (ht) | 4 | + | + | + | + | + | + | ? | + | + |
| Aguirre-Rodríguez et al. (2015) | c.565C>T (hm) | 3 | + | − | ? | ? | + | − | + | + | ? |
| Present case | c.379C>T (hm) | 5 | + | − | − | − | + | − | − | + | + |
Age: youngest patients described in families. +: present. −: not present. ?: not reported. hm: homozygosis. ht: heterocigosis.
Spastic paraplegia due to mutations in FA2H is an under diagnosed condition, and it should always be considered in childhood onset of progressive pyramidal dysfunction. An accurate genetic diagnosis is very important not only for treatment options and prognosis, but also for appropriate genetic counselling in parents and relatives regarding the recurrence risk within the family.
FundingThe authors declare no financial support.
Conflict of interestWe declare that no conflict of interest exists.
Thanks to Amelia Queipo, Begoña Rodriguez, Rebeca Moreno and Vanesa Barea for technical support.
