






La miotonía congénita es la forma más común de miotonía no distrófica. Esta miopatía está causada por mutaciones en el gen CLCN1, codificante del principal canal de iones cloruro del músculo esquelético (ClC-1); la alteración de la función de este canal, regulado por voltaje, da lugar al fenómeno de miotonía.
La enfermedad se puede heredar con un tipo de herencia dominante (enfermedad de Thomsen) o recesiva (enfermedad de Becker o miotonía congénita generalizada). El fenotipo clínico de ambas formas de la enfermedad es similar aunque la forma recesiva se caracteriza por una mayor gravedad de los síntomas.
El diagnóstico clínico de miotonía congénita debe sospecharse cuando encontramos en un paciente episodios de rigidez muscular (miotonía), remisión o alivio de la rigidez con el ejercicio (fenómeno warm-up), miotonía clínica, un patrón electromiográfico característico y/o historia familiar.
El diagnóstico molecular de miotonía congénita consiste en el análisis por secuenciación del gen CLCN1.
Myotonia congenita is the most common form of non-dystrophic myotonia. This myopathy is caused by mutations in the CLCN1 gene, encoding the main skeletal muscle chloride ion channel (ClC-1). Altering the function of this voltage-gated channel, leads to the phenomenon of myotonia.
The disease can be inherited with a dominant (Thomsen disease) or recessive type (Becker disease or congenital generalised myotonia). The clinical phenotype of both forms of the disease is similar, although the recessive form is characterised by more severe symptoms.
The clinical diagnosis of congenital myotonia should be suspected in a patient who presents with episodes of muscle stiffness (myotonia), remission or relief from stiffness with exercise (warm-up phenomenon), and a characteristic electromyography pattern, and/or family history.
Sequencing the CLCN1 gene is the present approach for molecular diagnosis of myotonia congenita.
La miotonía congénita (MC) es la forma más común de miotonía no distrófica1. Descrita por primera vez en el año 1876 por Julius Thomsen2,3, es una enfermedad producida por la alteración del principal canal de iones cloruro del músculo esquelético. Una reducción en la conductabilidad de Cl− del músculo lleva a un aumento de la excitabilidad de la célula muscular y produce el fenómeno de miotonía, un retraso en la relajación muscular después de una contracción voluntaria4.
Los individuos afectos de MC sufren rigidez muscular al iniciar el movimiento. Esta rigidez sin embargo remite después de varias repeticiones de ese mismo movimiento, dando lugar al fenómeno conocido como warm-up. El fenómeno miotónico no es exclusivo de la MC, ya que aparece en otras enfermedades como la parálisis periódica hiperpotasémica, la paramiotonía congénita, y las distrofias miotónicas 1 y 2, entre otras. Siendo la miotonía la manifestación clínica más llamativa de estas enfermedades es importante una atenta observación de los síntomas, una cuidadosa revisión de la historia clínica y del patrón de herencia para poder realizar un diagnóstico diferencial.
La MC es una enfermedad de causa genética que se produce por mutaciones en el gen CLCN15,6. La enfermedad puede ser heredada de manera autosómica dominante (enfermedad de Thomsen) o autosómica recesiva (enfermedad de Becker o miotonía generalizada recesiva, en inglés recessive generalized myotonia). El fenotipo clínico de estas dos formas de la enfermedad es similar y a menudo difícil de diferenciar y para ello debemos observar la intensidad de los síntomas de los pacientes y el patrón de herencia en la familia afectada (tabla 1).
Características de las dos formas clínicas de MC
| Enfermedad de Thomsen | Enfermedad de Becker | |
|---|---|---|
| Herencia | Autosómica dominante | Autosómica recesiva |
| Edad de aparición | Nacimiento | Primeros años |
| Músculos afectados | Grupos individuales de músculos | Aparición gradual que se generaliza a todos los músculos |
| Gravedad | Leve a moderada | Moderada a grave |
| Hipertrofia muscular | Ausencia a muy leve | Leve a evidente |
| Ratio de género | ♂ ≈ ♀ | ♂ ≈ ♀ |
La enfermedad de Thomsen (OMIM 160800) se manifiesta durante la infancia temprana. Se observa un retraso en la relajación muscular y, aunque no es frecuente observar hipertrofia muscular en niños afectos, sí que aparecen los músculos de las extremidades inferiores bien definidos. Habitualmente, la enfermedad de Thomsen no se identifica hasta la adolescencia, cuando se aprecia cierta torpeza y dificultad para el movimiento después del descanso. Tiene un espectro fenotípico de leve a moderado, siendo la expectativa de vida normal. Es frecuente encontrar heterogeneidad intrafamiliar referida a la edad de aparición, grupo muscular afectado y gravedad. No es raro diagnosticar la enfermedad en individuos clínicamente asintomáticos en los que la realización de un electromiograma demuestra un patrón miotónico.
La enfermedad de Becker o recessive generalized myotonia (OMIM 255700) es similar a la de Thomsen excepto que tiene un patrón de herencia autosómico recesivo y presenta generalmente un fenotipo más grave. Clínicamente se presenta durante la infancia o adolescencia precoz, acompañada a menudo de hipertrofia muscular. El fenómeno miotónico es más severo que en la enfermedad de Thomsen y los pacientes pueden experimentar periodos transitorios de debilidad al iniciar el movimiento7. Los individuos afectos pueden ver comprometido su control postural y sufrir caídas; también pueden producirse mialgias aunque no es muy común. Por último, a pesar de la hipertrofia muscular algunos individuos presentan atrofia de los antebrazos8,9.
PrevalenciaNo existen datos actualizados de la prevalencia de miotonía congénita y en un principio aún se aceptan las estimaciones de Becker de prevalencia entre 1:23000 y 1:5000010.
La forma recesiva de la enfermedad es más común que la dominante. Uno de los estudios en los que se apoyan estos datos es el de Fialho et al.11, en el que estudiaron una cohorte de 303 pacientes residentes en Reino Unido. Los resultados de frecuencia obtenidos por este grupo fueron de un 37% de herencia dominante frente a un 40% de herencia recesiva (familias o casos esporádicos con dos mutaciones). El resto (23%) corresponde a familias y casos esporádicos en los que solo fue detectada una mutación. En estos casos, la mutación encontrada estaba habitualmente asociada a herencia recesiva. Papponen et al.12 estiman una prevalencia para Finlandia de 7,3 en 100.000 habitantes, siendo preponderante la forma recesiva. En un estudio reciente en Inglaterra, se dan datos de muy baja prevalencia para miotonía congénita (0,52 en 100.000 habitantes)13.
Diagnóstico clínicoEl diagnóstico clínico de miotonía congénita se sospecha en los siguientes supuestos:
- a)
Episodios de rigidez muscular o calambres de aparición en la infancia.
- b)
Remisión o alivio de la rigidez con el ejercicio (fenómeno warm-up).
- c)
Contracción miotónica provocada por la percusión muscular. Esta se ensaya en músculos prominentes como la eminencia tenar, la lengua o el vasto lateral de cuádriceps (fig. 1)14.
 Figura 1.
Figura 1.Miotonía por percusión del muslo. Se utilizó un martillo de reflejos para golpear el extremo distal del músculo vasto lateral y esto produjo una contracción del músculo que persistió durante varios segundos.
Tomada de Lossin et al.14 con permiso del editor.
- d)
Patrón electromiográfico característico (fig. 2) que muestra descargas miotónicas: incrementos significativos de la actividad insercional y descargas repetitivas de elevada frecuencia. La duración de las descargas repetitivas se correlaciona con el retraso en la relajación muscular y representa la característica electrofisiológica típica de la enfermedad.
. Las hélices alfa (A a R) se dibujan como cilindros con la región extracelular hacia arriba y la región intracelular hacia abajo. Las dos mitades de la subunidad son de color verde y cian, y las regiones que forman la selectividad del filtro para Cl- se marcan en rojo. Tomada de Dutzler et al.24 con permiso del editor. El color de esta figura solo puede apareciarse en la versión electrónica del artículo.") Figura 2.
Figura 2.Estructura del monómero del canal ClC de Salmonella Typhimurium (StClC). Las hélices alfa (A a R) se dibujan como cilindros con la región extracelular hacia arriba y la región intracelular hacia abajo. Las dos mitades de la subunidad son de color verde y cian, y las regiones que forman la selectividad del filtro para Cl- se marcan en rojo.
Tomada de Dutzler et al.24 con permiso del editor. El color de esta figura solo puede apareciarse en la versión electrónica del artículo.
- e)
Historia familiar consistente con patrón de herencia autosómico dominante o recesivo. La historia familiar puede servir para distinguir entre estas dos entidades en algunos casos pero no siempre, debido a la variabilidad en la expresión de la enfermedad. Es siempre conveniente descartar la ausencia de miotonía en los progenitores mediante el EMG.
- f)
La biopsia muscular es poco informativa. Cuando se estudia el subtipo de fibras, se observa una deficiencia o ausencia completa del tipo 2B15.

. Las hélices alfa (A a R) se dibujan como cilindros con la región extracelular hacia arriba y la región intracelular hacia abajo. Las dos mitades de la subunidad son de color verde y cian, y las regiones que forman la selectividad del filtro para Cl- se marcan en rojo. Tomada de Dutzler et al.24 con permiso del editor. El color de esta figura solo puede apareciarse en la versión electrónica del artículo.")
El diagnóstico diferencial de MC incluye otros desórdenes genéticos en los cuales la miotonía es el hallazgo principal, tales como otras miotonías no distróficas: la parálisis periódica hiperpotasémica (OMIM 170500, 613345) y la paramiotonía congénita o enfermedad de Eulenburg (OMIM 168300). También es fundamental hacer el diagnóstico diferencial con las distrofias miotónicas tipo 1 y 2, DM1 y DM2 (OMIM 160900 y 602668, respectivamente).
La enfermedad conocida como paramiotonía congénita, al igual que la MC, es también una miopatía no distrófica causada por mutaciones en el gen SCN4A y puede a veces confundirse con la MC. Aunque ambos casos se presentan con debilidad generalizada desde la infancia, los individuos con paramiotonía tienen una sensibilidad extrema al frío que les provoca un empeoramiento de los síntomas. Además, en los individuos con paramiotonía la rigidez se agrava con la repetición de la contracción muscular, fenómeno contrario al warm-up observado en los pacientes con MC. Otra enfermedad provocada por mutaciones en el gen SCN4A es la denominada miotonía agravada por potasio, en el que la miotonía se asocia a episodios de parálisis periódica hiperpotasémica. Los dos fenotipos relacionados con el gen SCN4A presentan una herencia autosómica dominante y son el resultado de mutaciones que provocan un aumento de función del canal del sodio. Se estima que más de un 20% de los pacientes con sospecha de padecer MC tienen mutaciones en SCN4A16.
Aunque en la enfermedad de Becker se observa cierto grado de debilidad y desgaste muscular, el patrón es muy diferente en las distrofias miotónicas y además en estas últimas se observan con frecuencia otras manifestaciones extramusculares como las cataratas tempranas, alteración de la conducción cardiaca y la disfunción endocrina17. Es interesante reseñar que el fenómeno miotónico asociado a DM1 y DM2 se debe a la maduración anómala del ARN mensajero del gen CLCN118.
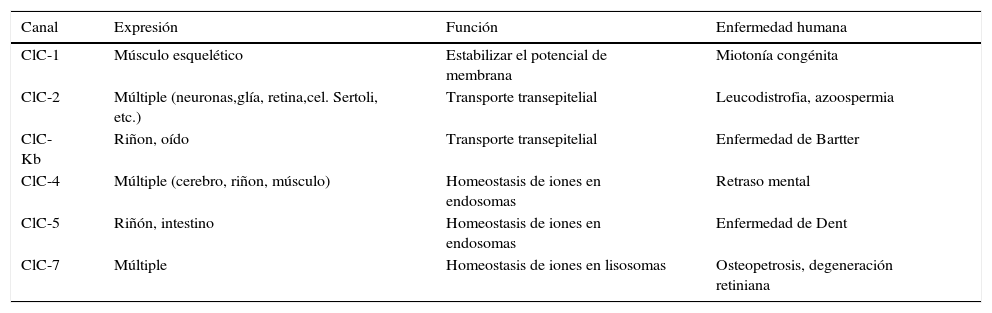
El canal ClC-1ClC-1 pertenece a la familia de canales iónicos regulados por voltaje, que en mamíferos se compone de nueve miembros19. Los canales de cloruro regulados por voltaje, canales «CLC», están presentes en todas las especies superiores y en la mayoría de los procariotas. En la especie humana, los canales CLC se clasifican en tres grupos atendiendo a su semejanza en la secuencia nucleotídica. El grupo de canales anclados a la membrana plasmática está formado por CLC-1 (expresión predominante en músculo esquelético), CLC-2 (expresión ubicua) y canales CLC-K (expresión predominante en riñón). Los otros dos grupos están formados por canales anclados a la membrana intracelular de organelas. La disfunción de algunos de estos canales provoca enfermedades en la especie humana (tabla 2).
Familia de canales ClC y su asociación con enfermedades humanas
| Canal | Expresión | Función | Enfermedad humana |
|---|---|---|---|
| ClC-1 | Músculo esquelético | Estabilizar el potencial de membrana | Miotonía congénita |
| ClC-2 | Múltiple (neuronas,glía, retina,cel. Sertoli, etc.) | Transporte transepitelial | Leucodistrofia, azoospermia |
| ClC-Kb | Riñon, oído | Transporte transepitelial | Enfermedad de Bartter |
| ClC-4 | Múltiple (cerebro, riñon, músculo) | Homeostasis de iones en endosomas | Retraso mental |
| ClC-5 | Riñón, intestino | Homeostasis de iones en endosomas | Enfermedad de Dent |
| ClC-7 | Múltiple | Homeostasis de iones en lisosomas | Osteopetrosis, degeneración retiniana |
Datos tomados y resumidos de la publicación de Jentsch et al.19.
Los canales iónicos regulados por voltaje son proteínas críticas para el establecimiento del potencial de reposo de la membrana celular y la propiedad de esta de generar potenciales de acción. En el funcionamiento normal del potencial de acción muscular los canales de sodio regulados por voltaje al abrirse generan el potencial de acción con la despolarización de la membrana de la fibra muscular. A continuación, y a la vez que se inactivan los canales de sodio, se produce la apertura de los canales de potasio regulados por voltaje y con ello la repolarización de la membrana. El canal de cloruro es responsable de la mayor parte de la polaridad de la membrana en reposo y así tiene la función de frenar la excitabilidad de la membrana y estabilizar el potencial de reposo9,20,21. La reducción de la conductabilidad del canal ClC-1, debido a mutaciones en el gen CLCN1 que lo codifica, tiene un efecto determinante sobre la excitabilidad de la fibra muscular. Las mutaciones en ClC-1 son mutaciones de pérdida de función que reducen por tanto la conductabilidad para iones cloruro y su papel estabilizador9.
Para entender los efectos patogénicos que producen las mutaciones en ClC-1 es necesario conocer algunas propiedades generales de los canales de cloro. Los canales ClC funcionan como homodímeros, en los que cada monómero (de 60-110kDa dependiendo del canal de que se trate) alberga un poro21. Cada poro individual (protoporo) del canal mantiene sus propiedades individuales, tales como selectividad iónica y conductabilidad, y cada protoporo puede abrirse o cerrarse de manera individual (apertura rápida). Además de la apertura individual, existe también un mecanismo de apertura común que abre ambos poros en paralelo (apertura lenta)22,23. Tanto la apertura del protoporo como la puerta común se activan con la despolarización20,24.
Cada subunidad de CLC-1 contiene 17 dominios α-hélice intramembana siendo el primero de ellos, dominio A, citoplasmático. Cada subunidad tiene una estructura interna que se repite y estas se sitúan en una disposición antiparalela, de forma que, la hélice B se corresponde con la hélice J, la hélice C con la hélice K y así sucesivamente (fig. 2). Además de estar unidas las dos mitades de la proteína mediante la unión I-J, ambas mitades de cada monómero están en contacto mediante las hélices C y K, H y P respectivamente24.
Los dos monómeros que forman el canal se disponen formando un ángulo de 45°, entre ellos existe una zona de interfase en la que se enfrentan e interaccionan ambas subunidades, formadas por las hélices H, P, I y Q24. Esta arquitectura del canal tiene importantes consecuencias en el efecto que producen las mutaciones en ClC-1 (fig. 3).

Descarga miotónica en un paciente con miotonía congénita tipo Thomsen. Registro continuo, barrido=1 seg. En reposo el músculo debería estar «en silencio», sin actividad. Sin embargo hay una actividad continua de descargas repetitivas a frecuencia descendente desde 120Hz hasta 50Hz, desapareciendo.
Los canales ClC carecen de un sensor de voltaje similar al que se ha descrito en otros canales iónicos dependientes de voltaje. La dependencia de voltaje para la apertura del protoporo se cree que resulta del movimiento de aniones a través del poro25,26, de modo que dicha apertura depende de la concentración de cloruros y el voltaje. El análisis por difracción de rayos-X de los canales ClC bacterianos reveló que en el lugar de unión del ion cloruro en el poro intervienen cuatro hélices provenientes de ambos lados de la membrana. Ha sido propuesto que un residuo de glutamato sobresaliente dentro del poro participa en el cierre/apertura del canal. De hecho, se ha demostrado que mutaciones en este residuo alteran de forma importante la apertura del canal27.
Mutaciones en el gen CLCN1El gen CLCN1, localizado en el brazo largo del cromosoma 7 (7q34), tiene un único tránscrito codificante (referencia EMBL: ENST00000343257, referencia en Genbank, NM_000083), distribuido en 23 exones, todos ellos codificantes; se compone de 3.172 nucleótidos y codifica una proteína de 988 aminoácidos.
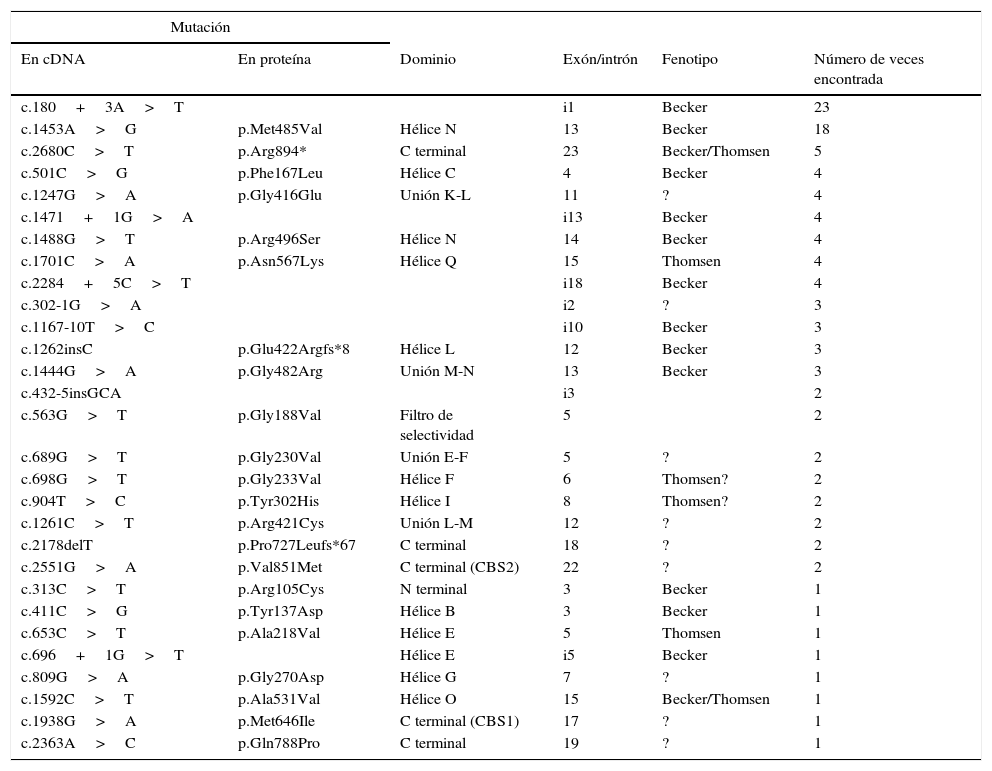
Hasta el momento se han descrito más de 200 mutaciones en CLCN1 según la base de datos Human Gene Mutation Database (www.hgmd.org), siendo en su mayoría mutaciones puntuales (missense, nonsense, pequeñas deleciones, inserciones e indels y mutaciones de splicing) y dentro de estas, las más abundantes son mutaciones de tipo missense/nonsense (200 mutaciones descritas). En la tabla 3, de mayor a menor frecuencia, se enumeran las mutaciones encontradas en el gen CLCN1 en una serie de 119 pacientes españoles de miotonía congénita. La mutación más frecuente en pacientes españoles es c.180+3A>T28, que es una mutación que altera la maduración del ARN (Escudero, comunicación personal de resultados no publicados).
Mutaciones identificadas en CLCN1 en una serie de pacientes españoles
| Mutación | |||||
|---|---|---|---|---|---|
| En cDNA | En proteína | Dominio | Exón/intrón | Fenotipo | Número de veces encontrada |
| c.180+3A>T | i1 | Becker | 23 | ||
| c.1453A>G | p.Met485Val | Hélice N | 13 | Becker | 18 |
| c.2680C>T | p.Arg894* | C terminal | 23 | Becker/Thomsen | 5 |
| c.501C>G | p.Phe167Leu | Hélice C | 4 | Becker | 4 |
| c.1247G>A | p.Gly416Glu | Unión K-L | 11 | ? | 4 |
| c.1471+1G>A | i13 | Becker | 4 | ||
| c.1488G>T | p.Arg496Ser | Hélice N | 14 | Becker | 4 |
| c.1701C>A | p.Asn567Lys | Hélice Q | 15 | Thomsen | 4 |
| c.2284+5C>T | i18 | Becker | 4 | ||
| c.302-1G>A | i2 | ? | 3 | ||
| c.1167-10T>C | i10 | Becker | 3 | ||
| c.1262insC | p.Glu422Argfs*8 | Hélice L | 12 | Becker | 3 |
| c.1444G>A | p.Gly482Arg | Unión M-N | 13 | Becker | 3 |
| c.432-5insGCA | i3 | 2 | |||
| c.563G>T | p.Gly188Val | Filtro de selectividad | 5 | 2 | |
| c.689G>T | p.Gly230Val | Unión E-F | 5 | ? | 2 |
| c.698G>T | p.Gly233Val | Hélice F | 6 | Thomsen? | 2 |
| c.904T>C | p.Tyr302His | Hélice I | 8 | Thomsen? | 2 |
| c.1261C>T | p.Arg421Cys | Unión L-M | 12 | ? | 2 |
| c.2178delT | p.Pro727Leufs*67 | C terminal | 18 | ? | 2 |
| c.2551G>A | p.Val851Met | C terminal (CBS2) | 22 | ? | 2 |
| c.313C>T | p.Arg105Cys | N terminal | 3 | Becker | 1 |
| c.411C>G | p.Tyr137Asp | Hélice B | 3 | Becker | 1 |
| c.653C>T | p.Ala218Val | Hélice E | 5 | Thomsen | 1 |
| c.696+1G>T | Hélice E | i5 | Becker | 1 | |
| c.809G>A | p.Gly270Asp | Hélice G | 7 | ? | 1 |
| c.1592C>T | p.Ala531Val | Hélice O | 15 | Becker/Thomsen | 1 |
| c.1938G>A | p.Met646Ile | C terminal (CBS1) | 17 | ? | 1 |
| c.2363A>C | p.Gln788Pro | C terminal | 19 | ? | 1 |
El diagnóstico genético de MC se establece en un probando cuando se detecta una mutación patogénica en heterocigosis en el caso de la enfermedad de Thomsen o una mutación en homocigosis o dos mutaciones en heterocigosis compuesta en el caso de enfermedad de Becker. El abordaje del diagnóstico molecular comienza por la secuenciación del gen CLCN1, seguida del estudio de grandes deleciones/duplicaciones mediante MLPA en caso de no identificar mutaciones patogénicas asociadas. Se estima que mediante la secuenciación del gen se pueden diagnosticar genéticamente más del 95% de los casos de miotonía congénita. La frecuencia de deleciones/duplicaciones en la población de pacientes de MC es entre el 1 y el 5% de los casos8,29.
La mayor parte de las mutaciones en CLCN1 se asocian a la forma recesiva de la enfermedad. Entre ellas están las mutaciones que generan proteínas truncadas incapaces de formar dímeros con los monómeros silvestres o que generan una ausencia de proteína debido a la disminución del ARNm por un deterioro del mismo (nonsense-mediated decay). Asimismo, las mutaciones que afectan aminoácidos que conforman el protoporo del canal, raramente pueden ejercer un efecto negativo sobre el otro protoporo en los dímeros mutante-silvestre, por lo que normalmente estarán relacionadas con miotonía de herencia recesiva. Otras mutaciones asociadas a la forma recesiva son las que impiden la correcta localización en la membrana celular de la proteína ClC-130.
Las mutaciones asociadas a miotonía congénita de la forma dominante son principalmente mutaciones missense. El efecto dominante se da en situaciones en las que una mutación afecta al cierre común del canal, o bien cuando el homodímero mutado provoca la disposición incorrecta o degradación del dímero resultante. Frecuentemente, el efecto de estas mutaciones se refleja en un cambio en el voltaje medio (V 0,5) en el que se produce la apertura del canal. De forma que, si el canal se abre solamente a voltajes más positivos que el potencial de membrana de la célula muscular, su efecto sobre la repolarización será nulo o estará muy disminuido31.
La mayor parte de las mutaciones con efecto dominante se localizan principalmente en las regiones que forman la interfase entre las 2 subunidades (hélices G, H, I, P y Q)32. La estructura dimérica del canal implica que, una mutación con efecto dominante negativo podrá disminuir la actividad del canal, como máximo, hasta el 25%. Esta es la fracción de dímeros silvestre-silvestre remanente cuando una mutación dominante está en heterocigosis. Este efecto parcial de las mutaciones dominantes explica por qué la miotonía congénita de Thomsen es generalmente menos grave y con menor penetrancia que la forma recesiva, en la cual ambos alelos están mutados y hay una pérdida total de función9.
Otras mutaciones en CLCN1 se han asociado a herencia dominante en algunas familias y a herencia recesiva en otras. Este fenómeno podría deberse a una expresión preferencial de uno de los alelos33. Por lo tanto, aunque se habla tradicionalmente de las formas dominante y recesiva, el límite entre estas dos formas de herencia no está totalmente definido.
TratamientoHistóricamente, la quinidina y la quinina se han utilizado como agentes antimiotónicos. Estos medicamentos son, por lo general, bien tolerados en dosis bajas. Sin embargo, la administración continuada de estos compuestos no está recomendada debido a su efecto tóxico que incluye alteraciones visuales y acústicas, vértigo y síntomas gastrointestinales. Además sus efectos antimiotónicos disminuyen con el uso repetido, de tal manera que el beneficio a largo plazo en el tratamiento de la miotonía es limitado.
Otros fármacos que se han utilizado en el tratamiento de la miotonía con distinto éxito incluyen la procaína, tocainida, mexiletina, carbamacepina, y fenitoína. Todos estos medicamentos actúan bloqueando los canales de sodio dependientes de voltaje. Mexiletina ha sido el medicamento de elección aunque su uso debe estar controlado porque existe un riesgo potencial proarritmogénico. La acetazolamina, un inhibidor de la anhidrasa carbónica, es el fármaco indicado en el tratamiento de la paramiotonía congénita, aunque a dosis más bajas ha sido utilizado también en niños con miotonía congénita34. Recientemente, Novak et al.35 han estudiado el efecto en ratones de dos fármacos, la lacosamida y la ranolazina, que provocan una inactivación lenta del canal de sodio. El efecto en ratones es superior al de la mexiletina pero todavía no se han realizado ensayos clínicos con estos fármacos.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a pacientes y familiares su disponibilidad para participar en el estudio. Gracias también a Rubén de Sancho y Blanca Ejarque por su contribución en la realización de los experimentos y a Rocío Mena y María Victoria Gómez por la secuenciación de los genes CLCN1 y SCN4A.
