




La trisomía del cromosoma 8, conocida como síndrome de Warkany, es una rara enfermedad genética que cursa con un fenotipo muy variable. Su principal característica clínica es la discapacidad intelectual, facies dismórficas y pliegues plantares profundos. Presentamos el caso de un paciente de 10 años de edad, con facies gargoloides, retraso mental y rigidez en las articulaciones. El estudio inicial del cariotipo, en el que se analizaron 20 metafases, fue normal. Se solicitó array de polimorfismos de nucleótido único (SNPs) al laboratorio.
ResultadosSe detectó una ganancia completa del cromosoma 8, que se interpretó como una trisomía 8 en mosaico de aproximadamente un 20%, y que era compatible con la clínica que presentaba el paciente.
DiscusiónEste caso muestra las limitaciones que tiene el análisis de solo 20 metafases en el cariotipo en pacientes con aneuploidías en mosaico. En estos casos estaría recomendado ampliar el estudio a al menos 30 metafases de cara a detectar mosaicismos en baja proporción.
Chromosome 8 trisomy, known as Warkany syndrome, is a rare genetic disease that has a very variable phenotype. Its main clinical characteristic is intellectual disability, dysmorphic facies, and deep plantar folds. The case is presented of a 10-year-old patient with gargoyle-like facies, mental retardation, and joint stiffness. The initial study of the karyotype, in which 20 metaphases were analysed, was normal. A single nucleotide polymorphisms (SNPs) array was requested from the laboratory.
ResultsA complete gain of chromosome 8 was detected, which was interpreted as a mosaic trisomy 8 of approximately 20%, and which was compatible with the clinical presentation of the patient.
DiscussionThis case shows the limitations of the analysis of only 20 metaphases in the karyotype in patients with mosaic aneuploidies. In these cases it would be recommended to extend the study to at least 30 metaphases in order to detect mosaicisms in low proportion.
La trisomía del cromosoma 8 en mosaico (T8M), conocida también como síndrome de Warkany, es una alteración cromosómica rara caracterizada por la presencia de 3 copias del cromosoma 8 en algunas células del organismo. Se produce como consecuencia de un evento poscigótico, normalmente por un error en la segregación de los cromosomas durante la mitosis en un feto con cariotipo normal, o por una corrección espontánea de una trisomía 8 total. La trisomía total del cromosoma 8 es normalmente letal y está provocada por una anomalía en la segregación de los cromosomas en la meiosis1.
La T8M tiene una prevalencia de 1:25.000-1:50.000 nacidos, siendo 5 veces más prevalente en hombres que en mujeres2. Tiene una gran variabilidad fenotípica que puede ir desde individuos sanos a otros con severas malformaciones. El fenotipo incluye facies anormales, alteraciones oculares, discapacidad intelectual, retraso en el crecimiento, disminución de la movilidad articular, clinodactilia, pliegues plantares profundos, enfermedades cardiacas, malformaciones renales, así como un mayor riesgo con respecto a la población pediátrica general de desarrollar leucemia y síndrome mielodisplásico. En ausencia de malformaciones graves, la esperanza de vida de estos pacientes es normal3.
El diagnóstico se basa habitualmente en la determinación del cariotipo. El asesoramiento genético es alentador, ya que la trisomía 8 es prácticamente siempre un incidente de novo, con un bajo riesgo de recurrencia.
Presentamos el caso de un paciente de 10 años con discapacidad intelectual y fenotipo peculiar, que fue diagnosticado, tras varios años de incertidumbre para sus familiares, de síndrome de Warkany.
Caso clínicoEl paciente fue fruto de la primera gestación de padres sanos y no consanguíneos, de 25 y 29 años de edad. La gestación se llevó a término sin incidencias, siendo un parto natural en el que el niño pesó 3.600g.
Con 3 años de edad, el paciente fue derivado al servicio de Pediatría por retraso psicomotor y retraso en la adquisición del lenguaje, tanto a nivel expresivo como a nivel comprensivo; en primer lugar se sospechó un retraso madurativo del paciente. Pesaba 12,3kg (P10) y presentaba una talla de 86,5cm (P3). En la exploración física se puso de manifiesto un cráneo dolicocefálico, frente abombada, implantación baja del cabello, orejas grandes que presentaban una implantación también baja, labio inferior grueso con paladar ojival, hiperlordosis, hipertricosis en la región lumbar, hernia umbilical y discreta rigidez articular. A nivel neurológico presentaba discreta hipotonía general, sialorrea constante y una marcha torpe. Se realizaron diversas pruebas complementarias: perfil bioquímico de alteraciones musculares, estudio de mucopolisacaridosis, muestreo de metabolitos orgánicos y cariotipo en sangre periférica, siendo todo normal, por tanto se descartaban enfermedades metabólicas que cursasen con la clínica del paciente. En el cariotipo se estudiaron 20 metafases, obteniendo un resultado cromosómico 46,XY.
A los 8 años de edad se realizó electromiograma para valoración de la conducción nerviosa, no detectándose ninguna alteración. Un año después se solicitó resonancia magnética nuclear (RMN), en la cual se objetivó un área de gliosis-encefalomalacia en la sustancia blanca peritrigonal y frontal que tenía un aspecto crónico. Para descartar enfermedad lisosomal se solicitaron nuevas pruebas complementarias: detección de glucosaaminoglicanos, oligosacáridos, sulfátidos en orina y linfocitos vacuolados, estando todo dentro de la normalidad.
Mediante un mapeo óseo y una telecolumna se evidenció una escoliosis. En la exploración física seguían observándose las características fenotípicas anteriores, más otras como clinodactilia leve del dedo meñique de la mano izquierda, leve engrosamiento de las articulaciones de las manos, lengua con tendencia a la protusión, caninos incisivos centrales prominentes, labio inferior grueso y algo evertido y una lesión hiperpigmentada de bordes mal definidos en el abdomen. A nivel neurológico, el paciente presentaba un ligero retraso mental con retraso en el desarrollo del lenguaje. Se decidió realizar array de polimorfismos de nucleótido único (SNPs) buscando alguna alteración genética que permitiera establecer un diagnóstico definitivo en el paciente.
Se realizó array de SNPs CytoScan® 750K, de Affymetrix®, a partir de ADN obtenido de leucocitos de sangre periférica. El análisis de los resultados se llevó a cabo con el software Chromosome Analysis Suite (ChAS) v.3.1.0.15 de Affymetrix® con el método CytoScan750K_Array Single Sample Analysis: NA33. Los filtros aplicados fueron 100Kb de tamaño mínimo para reportar variaciones en el número de copias y de 5Mb de tamaño mínimo para regiones con pérdidas de heterocigosidad. La referencia del genoma humano que se empleó fue NCBI37 (hg19). Se detectó una ganancia del cromosoma 8 completa, que se interpretó como T8M de aproximadamente el 20%, que confirmaba el diagnóstico de síndrome de Warkany (fig. 1). Una vez obtenidos estos resultados se repitió el cariotipo, en esta ocasión analizando 50 metafases, y se obtuvo la siguiente fórmula cromosómica: mos47,XY,+8[4]/46,XY[46]. Esto indicaba la presencia de un mosaicismo del cromosoma 8 en una proporción baja, de un 8%. Se realizó cariotipo a los padres analizando 50 metafases, la madre con una fórmula cromosómica 46,XX y el padre 46,XY, ambos normales. Todos los cariotipos se realizaron en leucocitos de sangre periférica.
Discusión
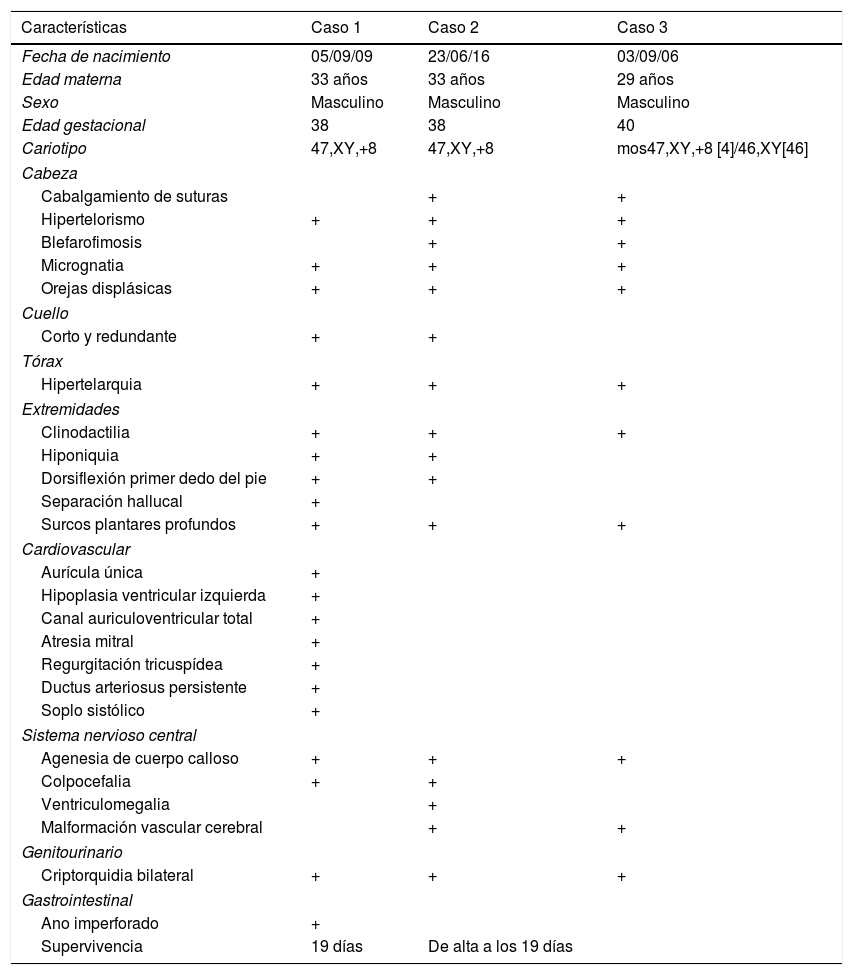
Los pacientes con T8M se caracterizan por la presencia de una dismorfia facial característica. No obstante, dado que es una entidad rara, el diagnóstico clínico puede ser difícil. A nivel craneofacial, los pacientes pueden presentar macrocefalia con frente alta y prominente, cara alargada, hipertelorismo, ptosis, estrabismo, nariz ancha, labio inferior grueso y evertido, retromicrognatia y orejas prominentes. El cuello suele ser corto y ancho, tórax con cifo-escoliosis dorso-lumbar, pecho en embudo y pelvis hipoplásicas. En las extremidades se puede observar clinodactilia y braquidactilia y son característicos los surcos palmo-plantares profundos. En niños se ha descrito criptorquidia, hipoplasia testicular y pubertad retrasada, así como oligospermia grave en la edad adulta. En el momento del nacimiento pueden no encontrarse anomalías, pero con la edad pueden aparecer deformidades graves en las articulaciones. Las malformaciones internas suelen ser raras, siendo las cardiacas y renales las más importantes. Neurológicamente los pacientes pueden presentar retraso mental, no destacando por su gravedad4. En la tabla 1 se hace una revisión del fenotipo del paciente comparado con el de otros 2 pacientes.
Comparativa fenotípica de 3 casos
| Características | Caso 1 | Caso 2 | Caso 3 |
|---|---|---|---|
| Fecha de nacimiento | 05/09/09 | 23/06/16 | 03/09/06 |
| Edad materna | 33 años | 33 años | 29 años |
| Sexo | Masculino | Masculino | Masculino |
| Edad gestacional | 38 | 38 | 40 |
| Cariotipo | 47,XY,+8 | 47,XY,+8 | mos47,XY,+8 [4]/46,XY[46] |
| Cabeza | |||
| Cabalgamiento de suturas | + | + | |
| Hipertelorismo | + | + | + |
| Blefarofimosis | + | + | |
| Micrognatia | + | + | + |
| Orejas displásicas | + | + | + |
| Cuello | |||
| Corto y redundante | + | + | |
| Tórax | |||
| Hipertelarquia | + | + | + |
| Extremidades | |||
| Clinodactilia | + | + | + |
| Hiponiquia | + | + | |
| Dorsiflexión primer dedo del pie | + | + | |
| Separación hallucal | + | ||
| Surcos plantares profundos | + | + | + |
| Cardiovascular | |||
| Aurícula única | + | ||
| Hipoplasia ventricular izquierda | + | ||
| Canal auriculoventricular total | + | ||
| Atresia mitral | + | ||
| Regurgitación tricuspídea | + | ||
| Ductus arteriosus persistente | + | ||
| Soplo sistólico | + | ||
| Sistema nervioso central | |||
| Agenesia de cuerpo calloso | + | + | + |
| Colpocefalia | + | + | |
| Ventriculomegalia | + | ||
| Malformación vascular cerebral | + | + | |
| Genitourinario | |||
| Criptorquidia bilateral | + | + | + |
| Gastrointestinal | |||
| Ano imperforado | + | ||
| Supervivencia | 19 días | De alta a los 19 días | |
Los casos 1 y 2 son descritos por Talavera Vargas Machuca et al.11; el caso 3 es el que se describe en este trabajo.
En el ámbito prenatal, el diagnóstico se puede sospechar ante la presencia de agenesia de cuerpo calloso, alteraciones cardiacas congénitas (presente en el 25% de los pacientes), malformaciones a nivel renal tales como reflujo uretral e hidronefrosis y también se han descrito malformaciones congénitas a nivel hepático como atresia biliar extrahepática y derivación portocava extrahepática. Algunos autores establecen la alfafetoproteína como uno de los marcadores en suero materno que está marcadamente elevado cuando hay una T8M. Por ello, una elevación importante de la alfafetoproteína y alteraciones ecográficas características pueden orientar hacia el diagnóstico de esta patología5. No obstante, la alfafetoproteína es una proteína que no se determina normalmente en las embarazadas, únicamente en el cribado bioquímico combinado del segundo trimestre. En nuestro paciente, no se solicitó el estudio de alfafetoproteína ni se detectaron anomalías ecográficas sugestivas.
En el laboratorio, la detección de trisomías en mosaico, como la T8M, puede ser todo un reto, ya que en la mayoría de los casos no hay sospecha de mosaicismo y por tanto las técnicas elegidas no son las correctas. Las técnicas de elección para detectar cromosomopatías en mosaico son el cariotipo y si se conoce o se sospecha el cromosoma implicado en la aneuploidía, la hibridación in situ fluorescente (FISH). La Asociación Europea de Citogenetistas (ECA) recomienda que cuando hay sospecha de alteraciones numéricas en mosaico, el número de metafases estudiadas debe ser suficiente para descartarlas, siendo 30 el mínimo. Cuando no exista tal sospecha, se recomienda para muestras posnatales el estudio de 20 metafases obtenidas a partir de leucocitos de sangre periférica6.
Además, el diagnóstico genético de T8M es complicado ya que normalmente la línea celular anormal, es decir, la portadora de la trisomía 8, tiende a desaparecer con la edad de las células sanguíneas7. En muchas ocasiones es necesario, ante la sospecha clínica, realizar el cariotipo de un cultivo de fibroblastos obtenido de una muestra de piel del paciente para confirmar el diagnóstico. Es importante recordar que los mosaicismos cromosómicos se relacionan con alteraciones en la pigmentación de la piel. Nuestro paciente presentaba una zona cutánea hiperpigmentada en el abdomen que podría haber hecho sospechar la presencia de alguna alteración genética en mosaico8.
En el caso que presentamos, el diagnóstico se realizó de manera accidental mediante array de SNPs de 750K, debido a que en nuestro laboratorio se estaba evaluando la efectividad de este tipo de array en muestras posnatales. Sin embargo, las recomendaciones sitúan el array CGH de 180K como la prueba de elección en pacientes con discapacidad intelectual y/o anomalías congénitas9. Los arrays de SNPs presentan la ventaja que permiten detectar mosaicismos de menor grado que los arrays de CGH, hasta de un 5%10. Esto es así debido a que las sondas de SNPs del array permiten obtener el genotipado (fig. 2).
, heterocigoto (AB) u homocigoto B (BB). El software de análisis asigna al genotipo A un valor de +0,5 y al B un valor de −0,5, por lo que las líneas se localizan en +1 (AA=0,5+0,5), 0 (AB=0,5−0,5) y −1 (BB=−0,5−0,5). En la muestra 1, las 3 líneas indican las combinaciones posibles que se corresponden con la existencia de 2 alelos, y por lo tanto, de 2 copias del cromosoma. En la muestra 2, la existencia de 4 líneas situadas en valores intermedios (entre +1,5 y +1; entre +0,5 y 0; entre 0 y −0,5; entre −1 y −1,5) indica la existencia de una trisomía en mosaico de, aproximadamente, el 50%. En la muestra 3, las 4 líneas indican las combinaciones diferentes que se corresponden con la existencia de una trisomía del cromosoma, siendo los posibles genotipos «A» o «B» como siempre para cada SNP y las posibles combinaciones: AAA, AAB, ABB y BBB. En este caso las líneas de sitúan en +1,5 (AAA=0,5+0,5+0,5), +0,5 (AAB=0,5+0,5−0,5), −0,5 (ABB=0,5−0,5−0,5) y −1,5 (BBB=−0,5−0,5−0,5).")
En el esquema de la parte superior de la figura, se representa cómo se sitúan las líneas de la gráfica del genotipado de SNPs a medida que aumenta el grado de mosaicismo de una trisomía. En el eje X, CN explica el número de copias, seguido del % que expera el % de mosaicismo. En el eje Y, la dotación cromosómica. Los posibles genotipos para cada SNP son «A» o «B» y las posibles combinaciones: homocigoto A (AA), heterocigoto (AB) u homocigoto B (BB). El software de análisis asigna al genotipo A un valor de +0,5 y al B un valor de −0,5, por lo que las líneas se localizan en +1 (AA=0,5+0,5), 0 (AB=0,5−0,5) y −1 (BB=−0,5−0,5).
En la muestra 1, las 3 líneas indican las combinaciones posibles que se corresponden con la existencia de 2 alelos, y por lo tanto, de 2 copias del cromosoma.
En la muestra 2, la existencia de 4 líneas situadas en valores intermedios (entre +1,5 y +1; entre +0,5 y 0; entre 0 y −0,5; entre −1 y −1,5) indica la existencia de una trisomía en mosaico de, aproximadamente, el 50%.
En la muestra 3, las 4 líneas indican las combinaciones diferentes que se corresponden con la existencia de una trisomía del cromosoma, siendo los posibles genotipos «A» o «B» como siempre para cada SNP y las posibles combinaciones: AAA, AAB, ABB y BBB. En este caso las líneas de sitúan en +1,5 (AAA=0,5+0,5+0,5), +0,5 (AAB=0,5+0,5−0,5), −0,5 (ABB=0,5−0,5−0,5) y −1,5 (BBB=−0,5−0,5−0,5).
No obstante, el array de CGH de 180K, que habría sido el recomendado en este caso, también hubiese podido detectar en teoría la T8M, ya que este tipo de array es capaz de detectar mosaicismos mayores de un 10%. Sin embargo, no fue posible comprobarlo.
Por último, resulta curiosa la discrepancia en el porcentaje de mosaicismo que se vio entre el cariotipo (8%) y el array (20%). Esto se puede deber a que el array permite estudiar todo el ADN de la muestra, que comprende células en todos sus estadios de división, mientras que con el cariotipo solo se visualiza el ADN de un número pequeño de células que se encuentran en metafase (en este caso, 50 células). Probablemente el grado de mosaicismo sería más similar si se hubiesen contado más células en el cariotipo o si se hubiera realizado FISH utilizando una sonda centromérica del cromosoma 8. Con el FISH también se analizan los leucocitos en todos los estadios de división, no solo los que están en metafase. La discrepancia observada también se puede deber al recambio propio de las células sanguíneas, que da lugar a que no siempre haya la misma proporción de células con la alteración genética en sangre periférica. La extracción de sangre para estos 2 estudios no se realizó el mismo día.
ConclusionesEl diagnóstico genético de las cromosomopatías en mosaico es complicado debido a que en muchas ocasiones no hay sospecha clínica, y por ello, no se elige en el laboratorio la metodología idónea para su detección.
Nuestro paciente fue diagnosticado de T8M mediante array de SNPs, que no es la prueba de elección para el diagnóstico de aneuploidías en mosaico, ya que mosaicismos de bajo grado pueden no ser detectados. Para diagnosticar pacientes con alteraciones numéricas en mosaico, la prueba de elección es el cariotipo, con la salvedad de que el número de metafases a estudiar debe ser mayor, recomendándose al menos el estudio de 30 metafases.
FinanciaciónLos autores declaran que no han recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún tipo de conflicto de intereses.
Agradecemos el tiempo dedicado a todos los profesionales que han contribuido en la elaboración del presente artículo.
