





El lupus eritematoso sistémico (LES) es un trastorno autoinmune con base genética, caracterizado por la aparición de autoanticuerpos, formación y depósito de complejos inmunes circulantes e inflamación crónica en varios órganos.
La etiología es multifactorial y, en individuos genéticamente predispuestos, factores medioambientales y componentes hormonales juegan un rol clave en el sistema inmune de esta enfermedad.
Cerca de 25 loci genéticos han sido identificados, indicando la importancia en esta enfermedad; sin embargo, la tasa de concordancia para el LES es de tan solo el 25% entre gemelos monocigotos1,2.
Un ejemplo de ello son las deficiencias de los componentes iniciales en la vía clásica del complemento sérico como el C1q, C2 o C4, que si bien es infrecuente, confieren susceptibilidad genética para el LES en una tasa del 30% en caso de deficiencia del C4 y de más del 90% para una deficiencia del C1q3.
Por otro lado, se demostró que el C1q inhibe a las células dendríticas plasmocitoides (CDP) en la secreción de interferón alfa (IFN-α), proporcionando así un nuevo enlace entre la deficiencia del complemento y la activación de la vía del IFN4.
Por ello, el IFN-α es considerado como un actor central en la patogénesis del LES, encontrándose concentraciones séricas altas en los brotes de esta enfermedad5.
En consecuencia, estos IFN ejercen efectos claves en la fisiopatología del LES, lo que sugiere que esta citoquina no solo posee un efecto a nivel del sistema inmune innato, sino también en las respuestas inmunes adaptativas.
Teniendo en cuenta estos hechos, se puede anticipar que las CDP, fuente principal de secreción de IFN, están involucradas en dicha enfermedad autoinmune.
En esta revisión nos centraremos en la participación de las CDP y del IFN en el LES6,7.
Systemic Lupus Erythematosus (SLE) is an autoimmune disorder with a genetic basis, and is characterised by the appearance of autoantibodies, the formation and deposition of circulating immune complexes, and chronic inflammation in various organs.
It is of multifactorial origin, and in genetically predisposed individuals, environmental factors and hormonal components play a key role in the immune system of the disease. About 25 genetic loci have been identified, indicating the importance in this pathology. However, the concordance rate for SLE is only 25% among monozygotic twins.
An example of this is the deficiencies of the initial components in the classical serum complement pathway such as C1q, C2 or C4, which, although rare, confer genetic susceptibility for SLE at a rate of 30% in the case of C4 deficiency, and more than 90% for C1q deficiency.
It was also demonstrated that C1q inhibits plasmacytoid dendritic cells (pDCs) in the secretion of interferon-alpha (IFN-α), thus providing a new link between complement deficiency and activation of the IFN pathway.
Therefore, IFN-α is considered to have a central role in the pathogenesis of SLE, with high serum concentrations being found in outbreaks of this disease.
These IFN exert prominent immunoregulatory effects, suggesting that this cytokine is key, not only in the innate immune system, but also in adaptive immune responses.
Taking these facts into account, it can be anticipated that pDCs, the main source of IFN secretion, are involved in this autoimmune disease.
In this review, we will focus on the participation of pDCs and IFNs in SLE.
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune cuya patogénesis es multifactorial, causada por factores genéticos, medioambientales y alteraciones del sistema inmunológico1–3.
La incidencia del LES es mayor en ciertos grupos étnicos, como los asiáticos, los latinoamericanos y los afronorteamericanos. La enfermedad sigue un curso crónico con periodos de remisión y exacerbación. Casi cualquier órgano puede verse afectado; su presentación es heterogénea, siendo desde una manifestación cutánea leve hasta manifestaciones del sistema nervioso central o del renal que ponen en peligro la vida4,5.
Varios genes se han asociado a la susceptibilidad del LES; cada uno de ellos presenta un pequeño efecto que sugiere esta asociación. Sin embargo, las interacciones entre el gen y el medio ambiente son todavía motivo de investigación.
Con respecto al sistema inmunológico, las células B han sido los primeros blancos terapéuticos en el LES; la importancia del sistema inmune innato y, en particular, las vías implicadas en la señalización del interferón (IFN) están emergiendo6. En la actualidad hay datos que apoyan un papel clave para las células dendríticas plasmocitoides (CDP), con un número de agentes terapéuticos biológicos e inhibidores de molécula pequeña que se encuentran en fase de investigación.
Datos recientes apuntan a formas alternativas de modulación de la vía del IFN y de los receptores de reconocimiento de patrones7,8.
Células dendríticasLas células dendríticas (CD) son las células presentadoras de antígenos más importantes que activan a las células T vírgenes y desempeñan funciones en las respuestas innatas frente a las infecciones y en forma conjunta entre el sistema inmune innato y el adaptativo.
Descubiertas a finales de los años 70 por Ralph Steinman y Cohn Zanvil9, estas células poseen una extraordinaria capacidad para las respuestas inmunitarias a antígenos extraños, actuando como verdaderos «centinelas» del sistema inmune.
Es importante mencionar que las CD se clasifican con base en distintos criterios como son: su localización en el organismo, su estado de madurez o su origen.
Clasificación de acuerdo con su localizaciónLas CD circulan a nivel sérico como células precursoras mieloides o linfoides, representando aproximadamente el 1% de las células mononucleares de sangre periférica (CMSP), en los tejidos no linfoides. A nivel de la piel se encuentran las CD epidérmicas, también conocidas como «células de Langerhans» (CL)10, mientras que en la dermis existen las «CD dermales»11, las cuales pertenecen a una subpoblación más amplia de «CD intersticiales»12, presentándose a nivel de la mayoría de los órganos, como son el hígado, los riñones, el corazón y otros tejidos conectivos. Las «CD asociadas a superficies mucosas» se encuentran en la mucosa de la cavidad oral, de los aparatos intestinal y respiratorio. Estas poblaciones de CD presentes en tejidos no linfoides actúan como células centinela captando antígenos en las barreras de entrada al organismo. Una vez que se han captado los antígenos, las CD migran hacia los órganos linfoides donde llevan a cabo la presentación de antígeno a los linfocitos T para que estos sean activados. Aunque el término CL se usa principalmente para referirse a las CD de la epidermis, este término se ha extendido a las CD presentes en todos los epitelios estratificados13. En los tejidos linfoides, el centro germinal, que es el microambiente que permite la generación de linfocitos B de memoria, también contiene «CD foliculares» y «CD de los centros germinales» (CDCG). Las CDCG son potentes células presentadoras de antígenos para los linfocitos T14,15.
Las CD que han capturado antígenos y que han migrado desde la piel, tejidos intersticiales no linfoides y de superficies mucosas hacia la linfa aferente, son reconocidas como «CD de linfa aferente», también llamadas «células veladas o veliformes»16. Estas células migrantes representan una fase intermedia entre las CL (CD inmaduras por excelencia) y las «CD interdigitantes» (CDI) en las que se transforman17. Estas últimas están presentes, principalmente, en los órganos linfoides secundarios, presentan un alto grado de maduración y pueden iniciar respuestas inmunes por activación de linfocitos T vírgenes. Al contrario de lo que sucede con las CL, las «CD del timo» (CD tímicas) parecen ser células no migrantes, que son generadas en el timo, donde completan su ciclo de vida18.
Clasificación por su estado de madurezLas CD han sido demostradas en distintos trabajos en los que se comparaban: CL aisladas en fresco («inmaduras») y CL procedentes de suspensiones epidérmicas posteriormente cultivadas in vitro («maduradas») revelaron sus distintas capacidades de captación y procesamiento antigénico y de estimulación de linfocitos T19. A modo de definición, las CD inmaduras se caracterizan por presentar una elevada capacidad fagocítica y de procesamiento antigénico, localizarse principalmente en regiones periféricas del organismo como la piel y mucosas y presentar una menor cantidad de moléculas del complejo mayor de histocompatiblidad (CMH-II) y de moléculas coestimuladoras. De manera opuesta, las CD maduras se dirigen a las zonas T de los órganos linfoides secundarios, donde queda reflejada su capacidad para la presentación de antígenos a los linfocitos T, siendo su actividad fagocítica más limitada. Además, sobreexpresan la molécula CMH-II superficialmente (y no en el citoplasma como en las inmaduras) y moléculas coestimuladoras20. Estas definiciones se ajustan al modelo clásico en el que las CD inmaduras presentes en piel y mucosas maduran durante su migración a los nódulos linfáticos, disminuyendo su capacidad de captación de antígenos y aumentando su capacidad de estimular a los linfocitos T21.
Clasificación de acuerdo con el origenSistema hematopoyético y mesenquimalLas CD de origen hematopoyético se dividen a su vez en CD procedentes de progenitores linfoides y mieloides22,23. Cuando se intenta clasificar las CD con base en su origen y funcionalidad, se hace uso de una amplia terminología que a veces puede ser muy complicada y ambigua. Este es el caso de acepciones como «mieloides», «linfoides», «convencionales», «plasmocitoides», «CD tipo1», «CD tipo2», «CDPI» (CD productoras de IFN), complicándose aún más debido a la diferente subdivisión que se hace de las CD cuando se habla de modelos murinos o de humanos, que son las 2 especies más estudiadas. Por todo ello, la forma más común de clasificar a las CD es aquella que las divide en convencionales (de origen principalmente mieloide) y en plasmocitoides (de origen principalmente linfoide)24.
Las CDP se identificaron por primera vez en áreas paracorticales de nódulos linfáticos reactivos, con una morfología similar a la de las células plasmáticas y con marcadores comunes a linfocitos T y monocitos. Poseen proyecciones membranarias largas y con capacidad fagocítica, distribuidas ampliamente en los tejidos linfáticos, el epitelio mucoso y el parénquima de los órganos25. Se observó también que estas células producían grandes cantidades de IFN de tipo 1 en respuesta a ciertos virus y que en determinadas condiciones de cultivo (IL3, CD40L) se transformaban en células de morfología estrellada y con capacidad para activar linfocitos T vírgenes. Por todo ello, los linfocitos T plasmocitoides, monocitos plasmocitoides o células productoras de IFN natural resultaron ser el mismo tipo celular denominado CDP26-28.
Y las de origen mesenquimal, cuya base son las CD foliculares, se diferencian del resto de las CD por ser células estromales típicas de tejido conectivo y lo único que comparten en común con las de origen hematopoyético es la morfología dendrítica. En este sentido, las CD foliculares no son verdaderas presentadoras de antígenos ya que no expresan moléculas del CMH-II y, por tanto, no presentan antígenos procesados a los linfocitos T CD4. En su lugar, presentan antígenos completos en forma de complejos inmunes a los linfocitos B, ayudando a su activación o maduración selectiva y contribuyendo en la formación de los centros germinales, de células plasmáticas y de linfocitos B de memoria29,30.
La disfunción de la apoptosis y su relación con las CD conduce a la liberación de autoantígenos, que inician una respuesta inmunológica; por lo tanto, se piensa que es una de las bases fisiopatológicas del LES31. Las CD reconocen y procesan antígenos para su presentación a los linfocitos T CD4, y en modelos animales se demostró que la pérdida de la autotolerancia conduce a la hiperactividad de las células T CD4 y, finalmente, al desarrollo de la enfermedad autoinmune. La fagocitosis del producto final de la apoptosis conduce a la maduración de las CD mieloides (CDM) y a la producción de citoquinas proinflamatorias, incluyendo la IL-632. En presencia de IL-6 y otras citoquinas proinflamatorias, las CDM maduras pueden inducir la activación de células Th1, Th2 y Th17, mientras que la IL-6 no permite la diferenciación de las células T vírgenes a células T reguladoras (Treg) en el LES33.
La familia de los interferonesHay 3 tipos de IFN conocidos hoy en día (IFN I, IFN II, e IFN III). Quizá el más estudiado y mejor conocido es el IFN I, cuya principal célula secretora son las CDP. Estos IFN I, todos con una homología estructural considerable, son codificados por genes situados en el cromosoma 9. Los IFN I más importantes en la defensa frente a los virus son el interferón alfa (IFN-α) (que en realidad abarca 13 proteínas diferentes muy relacionadas) y el interferón beta (IFN-β), que es una sola proteína34,35.
Los IFN I, y en particular IFN-α, han surgido como citoquinas patógenas clave en el LES. Sus funciones son: antivíricas, antiproliferativas y con efectos inmunomoduladores36. La señalización a través del IFN I se inicia tras la unión con el receptor de IFN (IFNAR): un complejo receptor que consta de 2 proteínas transmembrana, IFNAR1 e IFNAR2, que junto a la unión con 2 tirosina-quinasas citoplasmáticas, JAK1 y TYK2, permite la fosforilación de las 2 proteínas (STAT 1 y STAT 2); sumado a ello, el IRF9 forma en conjunto el llamado «complejo del factor de transcripción» ISGF3, que finalmente estimula en el núcleo de factores genéticos para la formación de IFN37 (fig. 1).
 con la transducción de señales mediada por la activación de JAK/STAT. Posterior a ello se producen mecanismos que dan como resultado la trascripción de genes para la producción de IFN. Fuente: adaptado de Amezcua-Guerra et al.66.")
Los 3 tipos de IFN con sus respectivas vías de señalización intracelular. Los IFN tipos I, II y III se acoplan a través de receptores distintos (IFNAR, IFNGR y IFNLR, respectivamente) con la transducción de señales mediada por la activación de JAK/STAT.
Posterior a ello se producen mecanismos que dan como resultado la trascripción de genes para la producción de IFN.
Fuente: adaptado de Amezcua-Guerra et al.66.
El IFN I induce una variedad de efectos biológicos que pueden aumentar la autoinmunidad a través de la alteración de la función de las células efectoras clave, tales como células B, células T y CD. Por ejemplo, in vitro, el IFN-α promueve la diferenciación de las células B autorreactivas y regula la secreción del «factor activador de las células B» (BAFF) y «factor de proliferación de células B» (APRIL, por sus siglas en inglés), lo que permite la activación, diferenciación y proliferación de las células B38. En algunos estudios, los IFN I inducen directamente la activación de las CDP mediante la expresión del CMH-II, y marcadores coestimuladores como los CD 40, CD 80, CD 86 y la producción de varias citoquinas que estimulan la diferenciación de monocitos y CD inmaduras en células presentadoras de antígenos eficaces39. El IFN-α también induce la diferenciación de células T vírgenes en células T cooperadoras31. Además, el IFN-α provoca la inactivación de las Treg mediante la regulación de AMP intracelular y la regulación de la señalización de receptores de células T, y estimula la generación de células T en los ganglios linfáticos foliculares residentes40.
En el LES, los niveles elevados de IFN-α están íntimamente asociados con manifestaciones de la enfermedad, incluyendo la producción de los autoanticuerpos (por ejemplo: anti-ADN) y manifestaciones renales, hematológicas y del sistema nervioso central41. En un modelo experimental BXSB, que produjo una disminución transitoria de las CDP, dio lugar a la mejoría clínica, que coincidió con la reducción de la transcripción genética de los IFN, sobre todo del IFN-α42. Efectos protectores similares se observaron también en el factor de transcripción TCF4, que carecía del toll-like receptor(TLR) 7, y en modelos de lupus B6.Sle1.Sle3 en el que las CDP fueron inhibidas funcionalmente mediante la supresión del factor de transcripción génica E2-243.
En conjunto, una activación continua del sistema de IFN I forzado por factores genéticos e inmunológicos en el LES conducirá a una respuesta autoinmune y a una inflamación crónica.
El papel de los toll-like receptor (TLR)Una creciente evidencia apoya la idea de que la activación de los TLR juega un rol importante en el mantenimiento y la progresión de la enfermedad. Los TLR7 y TLR9 son particularmente relevantes para el LES, y la estimulación a través de estos receptores conduce a niveles muy altos de producción de IFN-α. Microrganismos exógenos que actúan a través de estos TLR exacerban la enfermedad, y esto es consistente con la asociación observada en brotes de la enfermedad establecida con infecciones virales. Estos circuitos son, por lo tanto, inductores endógenos de IFN-α, y la producción resultante podría perpetuar el proceso autoinmune37,44.
Los estímulos más potentes para la síntesis del IFN I son los ácidos nucleicos virales. Por lo tanto, los receptores de tipo rig-like receptors (RIG), y los TLR 3, 7, 8 y 9 en las vesículas endosómicas, reconocen ácidos nucleicos víricos e inician vías de transmisión de señales que activan a la familia de factores de transcripción del IFN.
El factor de diferenciación mieloide 88 (MyD88) es una proteína presente en la mayoría de las vías de señalización de los TLR45. Debido a que tanto TLR7 como TLR9 utilizan esta proteína, MyD88 es un objetivo para el tratamiento en el LES. La inhibición del MyD88 en ratones muestra una disminución ostensible de enfermedades autoinmunes como, por ejemplo, la nefropatía lúpica46. Teichmann et al. investigaron más a fondo el mecanismo patogénico de MyD88 en diferentes tipos de células, revelando que MyD88 regula a las células B47.
Otro objetivo potencial en la inhibición de la señalización de TLR en el LES son las quinasas asociadas con IL-1R (IRAK) IRAK1 e IRAK4, que son activadas por el MyD88 y a su vez activan al TRAF6 en la señalización de TLR. Todos los TLR, excepto TLR3, requieren IRAK y MyD88 para su señalización48. Los pacientes con deficiencia de IRAK4 o MyD88 no muestran producción de autoanticuerpos en sangre y no desarrollan, por lo tanto, enfermedades autoinmunes, lo que sugiere que la inhibición de estas vías es esencial en la patogénesis del LES (fig. 2).
 utilizan al MyD88 como vía para la transducción de señales, lo que resulta en la producción de IFN I y citoquinas inflamatorias. Algunos anticuerpos u oligonucleótidos de molécula pequeña están siendo motivo de investigación para inhibir dichas vías. Fuente: adaptado de Wu et al.61")
Potenciales objetivos en la vía de los TLR en el LES. Los TLR (excepto el TLR3) utilizan al MyD88 como vía para la transducción de señales, lo que resulta en la producción de IFN I y citoquinas inflamatorias. Algunos anticuerpos u oligonucleótidos de molécula pequeña están siendo motivo de investigación para inhibir dichas vías.
Fuente: adaptado de Wu et al.61
Uno de los objetivos es la inhibición terapéutica con anticuerpos monoclonales enfocados en el IFN-α. Sifalimumab, un anticuerpo monoclonal anti-IFN-α, fue evaluado, y tras su administración produjo una disminución en los niveles del IFN, que fue más marcada en los pacientes que se encontraban en fase I del estudio clínico con un SLEDAI mayor de 5 (moderado) que en los pacientes en fase II con un SLEDAI mayor de 11 (severo)49,50. Se observó una franca respuesta clínica en las manifestaciones de piel y de articulaciones.
Se observó eficacia clínica en aquellos pacientes con valores séricos bajos de IFN-α, lo que sugiere quizás que se puede lograr una neutralización más eficaz de la actividad de IFN-α en pacientes con manifestaciones clínicas importantes pero con relativamente baja producción de este IFN51. Cabe destacar que todos los pacientes que habían recibido dosis únicas o múltiples de rontalizumab (otro tipo de anticuerpo monoclonal anti-IFN-α) recuperaron los niveles de IFN, 6 meses después de la última dosis.
Una estrategia interesante ha sido reportada en estudios preclínicos y más recientemente en un estudio en humanos del llamado IFN-kinoide, un complejo recombinante de IFN-α52. La inyección del IFN-kinoide dio como resultado una producción dependiente de células T de anticuerpos neutralizantes del IFN-α específicos que, a su vez, disminuyeron la expresión de genes estimulantes de IFN I52.
En vista de las sugerencias de que el IFN-α y el IFN-β podrían contribuir a la patogénesis del LES, el bloqueo del IFNAR podría proporcionar una terapia más eficaz para la enfermedad autoinmune sistémica que aquellos anticuerpos monoclonales que solo se dirigen a un subtipo (fig. 1).
La enfermedad observada en ratones con lupus BXSB que reciben un anticuerpo específico para IFNAR, junto con datos de ratones neozelandeses genéticamente deficientes en IFNAR, apoyan este enfoque terapéutico53. Un anticuerpo monoclonal específico para la subunidad 1 de IFNAR, MEDI-546, previene la asociación de la subunidad 1 de IFNAR con la subunidad 2, bloqueando así sucesivos procesos de señalización. En contraste con los efectos parciales sobre la expresión génica estimulada por IFN-α en los estudios de sifalimumab y rontalizumab, este medicamento anti-IFNAR dio como resultado una inhibición casi completa de la expresión de genes estimulantes de IFN (ISGs, por sus siglas en inglés) en sangre periférica54. Esta inhibición más completa de la vía del IFN I debe llevarse a cabo con precaución, teniendo en cuenta el papel central del IFN-α en la defensa del huésped durante la fase aguda de la infección viral.
La hidroxicloroquina (HCQ), medicamento usado contra la malaria, es un antagonista de los TLR 7, 8, 9. La actividad de la HCQ se ha atribuido a la reducción de la acidificación endosomal, que se requiere para la activación de TLR. Evidencias más recientes sugieren que la HCQ se une directamente a los ácidos nucleicos, causando modificaciones estructurales.
Numerosos agentes terapéuticos en desarrollo dirigen su atención a los TLR, o a sus moléculas, incluyendo oligonucleótidos e inhibidores de molécula pequeña. Varios oligonucleótidos actúan como antagonistas de los TLR. El estudio de la droga, DV-1179, un antagonista dual de TLR 7/9, fue evaluado y tolerado en fase I de investigación, pero no se evidenció una reducción de los genes reguladores del IFN55.
Estudios preclínicos con otro antagonista dual de TLR 7/9, el IRS-954, mostraron inhibición de la producción de IFN en las CDP en respuesta a los virus de ADN/ARN, disminución de los complejos inmunes circulantes y eficacia en modelos murinos56.
Curiosamente, un aumento de la sobrevida de las CDP mediada por TLR 7 y TLR 9 también fue revertido con el tratamiento del IRS-954 en ratones con lupus57.
Otro compuesto, OMI-3100, demostró que no solo inhibe al IFN-α sino también a la IL-17 a partir de células mononucleares de sangre periférica (CMSP)58.
Un antagonista TLR 7, 8 y 9, llamado OMI-8400, mostró una eficacia en modelos animales. Y está actualmente en fase I para el LES27,28. Tanto OMI-3100 como OMI-8400 han sido bien tolerados y, curiosamente, fueron eficaces en ensayos de fase II en la psoriasis, otra enfermedad autoinmune asociada al IFN59,60.
Los inhibidores de molécula pequeña tienen la ventaja potencial de su disponibilidad oral, y los compuestos han sido diseñados para apuntar a los TLR y proteínas de señalización de esta vía, como el MyD88. El derivado de quinazolina, CpG-52364, un inhibidor de molécula pequeña de TLR 7, 8 y 9, ha demostrado ser seguro y más eficaz que la HCQ en los estudios preclínicos con animales61,62, que han completado la fase I en ensayos clínicos en el LES, aunque no se han reportado aún resultados63. El inhibidor de la dimerización de MyD88, ST-2825, interfiere con la activación de IRAK 4 y IRAK 1 a través de MyD88, e inhibe la producción de citoquinas proinflamatorias y la proliferación de células B inducida por TLR 9 y su diferenciación64,65(tabla 1).
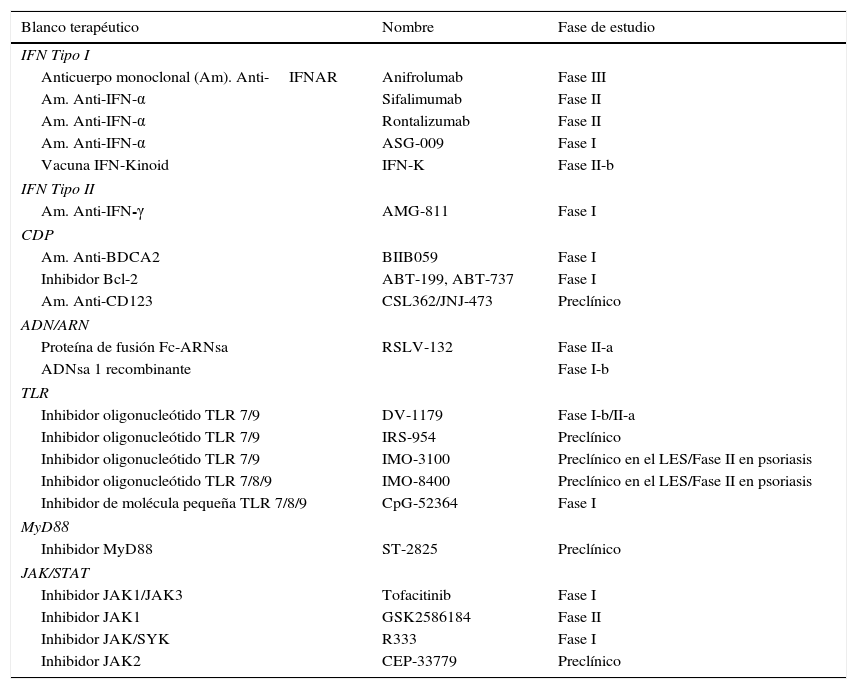
Objetivos terapéuticos de la vía del interferón en el LES
| Blanco terapéutico | Nombre | Fase de estudio |
|---|---|---|
| IFN Tipo I | ||
| Anticuerpo monoclonal (Am). Anti-IFNAR | Anifrolumab | Fase III |
| Am. Anti-IFN-α | Sifalimumab | Fase II |
| Am. Anti-IFN-α | Rontalizumab | Fase II |
| Am. Anti-IFN-α | ASG-009 | Fase I |
| Vacuna IFN-Kinoid | IFN-K | Fase II-b |
| IFN Tipo II | ||
| Am. Anti-IFN-γ | AMG-811 | Fase I |
| CDP | ||
| Am. Anti-BDCA2 | BIIB059 | Fase I |
| Inhibidor Bcl-2 | ABT-199, ABT-737 | Fase I |
| Am. Anti-CD123 | CSL362/JNJ-473 | Preclínico |
| ADN/ARN | ||
| Proteína de fusión Fc-ARNsa | RSLV-132 | Fase II-a |
| ADNsa 1 recombinante | Fase I-b | |
| TLR | ||
| Inhibidor oligonucleótido TLR 7/9 | DV-1179 | Fase I-b/II-a |
| Inhibidor oligonucleótido TLR 7/9 | IRS-954 | Preclínico |
| Inhibidor oligonucleótido TLR 7/9 | IMO-3100 | Preclínico en el LES/Fase II en psoriasis |
| Inhibidor oligonucleótido TLR 7/8/9 | IMO-8400 | Preclínico en el LES/Fase II en psoriasis |
| Inhibidor de molécula pequeña TLR 7/8/9 | CpG-52364 | Fase I |
| MyD88 | ||
| Inhibidor MyD88 | ST-2825 | Preclínico |
| JAK/STAT | ||
| Inhibidor JAK1/JAK3 | Tofacitinib | Fase I |
| Inhibidor JAK1 | GSK2586184 | Fase II |
| Inhibidor JAK/SYK | R333 | Fase I |
| Inhibidor JAK2 | CEP-33779 | Preclínico |
El autor declara no tener ningún conflicto de interés
