








El síndrome de Schnitzler es una enfermedad autoinflamatoria rara, sistémica, caracterizada típicamente por la presencia de urticaria crónica y gammapatía monoclonal. Se han reportado alrededor de 300 casos en todo el mundo desde su descubrimiento, siendo más común en Europa que en otras regiones como Norteamérica, Japón o Latinoamérica. Hasta el momento, no se han identificado factores de riesgo ni se ha observado un patrón de herencia relacionado con esta enfermedad. Sin embargo, se ha planteado la alteración en la vía de la interleucina 1 como mecanismo fisiopatológico principal. La complicación más habitualmente descrita asociada al trastorno es la transformación en linfoproliferación maligna o desarrollo de amiloidosis secundaria. El tratamiento consiste, principalmente, en el bloqueo de interleucina 1, con lo cual se ha demostrado un significativo control de los síntomas clínicos. En esta revisión buscamos dar una mirada profunda a los posibles mecanismos fisiopatológicos de esta rara entidad.
Schnitzler syndrome is a rare autoinflammatory systemic disease, whose characteristic features are typically chronic urticaria and monoclonal gammopathy. Over 300 cases have been reported worldwide since its discovery, more frequently in Europe than other regions such as North America, Japan, and Latin America. No predisposing conditions or inheritance pattern associated with this disease have been identified to date. However, it seems that disruption of interleukin 1 homeostasis plays an important role as the main pathophysiological mechanism. The main complications described associated with this disease are malignant lymphoproliferative disorders or the development of amyloidosis. Treatment consists primarily of interleukin 1 blockade and this has shown significant control of clinical symptoms. In this review we aim to take an in-depth look at the possible pathophysiological mechanisms of this rare entity.
El síndrome de Schnitzler (SchS) es una enfermedad sistémica rara, de carácter autoinflamatorio, clasificada como una inflamosomopatía adquirida, la cual comparte algunas características con los síndromes autoinflamatorios hereditarios [1]. Se reportó por primera vez en 1972 y fue descrita posteriormente, en 1974, por Liliane Schnitzler, dermatóloga francesa, como una constelación de síntomas y signos clínicos asociados con gammapatía monoclonal [2].
En la actualidad se desconoce la prevalencia exacta de esta enfermedad, sin embargo, desde su primera descripción en 1972 se han publicado cerca de 350 casos en el mundo, incluyendo Norteamérica y Japón, pero más frecuentemente en Europa. En Latinoamérica hay muy pocos casos informados y en Colombia se la considera como una enfermedad huérfana [3].
Se ha observado una ligera predominancia en el sexo masculino, con una relación hombre:mujer de 1,5:1 [4]. Los datos demográficos indican con claridad que el SchS es un trastorno que aparece en la edad adulta, con un pico en la sexta década de la vida, edad promedio a los 51 años. Dada su baja prevalencia y sospecha clínica, hay un retraso en la confirmación diagnóstica de 2 a 13 años (media de 6,1 años) después del inicio de los síntomas [5].
Hasta la fecha no se han identificado factores de riesgo ni hay datos que evidencien un patrón de herencia. La principal complicación, descrita en un 15-20% de los casos, es la progresión a un trastorno linfoproliferativo en particular, la enfermedad de Waldenström, y, de forma menos eventual, el desarrollo de amiloidosis AA (subtipo de amiloidosis que se caracteriza por la agregación y el depósito de fibrillas amiloideas compuestas por proteína amiloide A sérica) [6]. Con menos frecuencia se han asociado otros trastornos, como linfoma linfoplasmocítico, mieloma múltiple, leucemia linfocítica crónica, linfoma esplénico de la zona marginal y linfoma de células B de la zona marginal [7].
Este escrito tiene como objetivo hacer una revisión narrativa amplia y profunda de la literatura relacionada con el SchS, abarcando epidemiología, características clínicas, fisiopatología y tratamiento.
MétodosBúsqueda de la literaturaSe realizó una búsqueda bibliográfica desde agosto hasta septiembre de 2024 en las bases de datos PubMed y Google Scholar. Los artículos incluidos en esta revisión narrativa se identificaron mediante búsquedas bibliográficas de publicaciones en la base de datos PubMed, utilizando términos o palabras clave MeSH como «síndrome de Schnitzler» y «Schnitzler Syndrome» [MeSH]. Además, se hicieron búsquedas combinadas como: «Síndrome de Schnitzler y patogénesis», «Síndrome de Schnitzler y diagnóstico», «Síndrome de Schnitzler y tratamiento». Se tomaron artículos relevantes, sin límite de fecha de publicación, tanto estudios originales como revisiones de la literatura, que incluyeran información relacionada con esta afección.
Criterios de elegibilidadLos artículos fueron recopilados en una base de datos que incluía las palabras clave en el título o en el abstract en cualquier idioma. Se encontraron 95 artículos, de los cuales se excluyeron 16 duplicados o sin acceso a texto. Posteriormente a la revisión y exclusión de artículos con información redundante, se incluyeron en total 62 artículos, de los cuales 27 correspondían a revisiones narrativas, 33 a artículos originales (incluyendo estudios observacionales y estudios experimentales) y 19 a reportes de casos. Con los artículos seleccionados se hizo una síntesis de la información acerca de la epidemiología, las manifestaciones clínicas, los criterios diagnósticos, la fisiopatología y el tratamiento del SchS. En la figura 1 se muestra el proceso de selección de artículos para esta revisión narrativa.
Fisiopatología
Los trastornos autoinflamatorios sistémicos comprenden un grupo cada vez más amplio de enfermedades caracterizadas por la disfunción del sistema inmunológico innato y una importante estimulación de las vías inflamatorias, sin la participación de títulos elevados de autoanticuerpos ni células T específicas de antígeno [8]. Estos trastornos pueden ser de tipo monogénico, en los que se puede identificar una variante genética específica, principalmente debido a errores en el mecanismo inmunológico innato, o poligénicos, en los que no existe una única variante genética identificada y se observa la participación de múltiples genes; por tanto, su caracterización es compleja. El SchS pertenece al grupo de las inflamosomopatías complejas y adquiridas [9]. A continuación se describen algunos factores desencadenantes identificados en la fisiopatología de este trastorno.
Implicaciones genéticasLa gran similitud entre el SchS y los síndromes periódicos asociados a criopirina (CAPS) ha puesto sobre la mesa la posibilidad de que tengan mecanismos fisiopatológicos similares; ambas enfermedades comparten manifestaciones como fiebre, erupción similar a urticaria, inflamación que afecta a muchos sistemas de órganos, riesgo de desarrollo de amiloidosis AA y una respuesta significativa al tratamiento con antagonistas de la interleucina (IL)-1 (anti-IL-1) [10].
En CAPS se tiene conocimiento de la participación del gen NLRP3, el cual codifica componentes importantes en el inflamosoma [11]. NLRP3 es un receptor citosólico de reconocimiento de patrones intracelular que reconoce patrones moleculares asociados a patógenos y patrones moleculares asociados a daño; tras la estimulación, NLRP3 selecciona la proteína adaptadora tipo speck asociada a apoptosis y la procaspasa 1, y forman el inflamosoma para activar la caspasa 1. De esta manera, se inicia el procesamiento de la IL-1β y de la IL-18 para producir citocinas maduras [12].
Las mutaciones por ganancia de función del gen NLRP3 se relacionan con un aumento de la producción de IL-1β e IL-18, que son potentes moléculas proinflamatorias [11]. La posibilidad de la participación en la fisiopatología de la activación del inflamosoma ha impulsado a los investigadores a relacionar el SchS con alguna mutación o variante patológica; sin embargo, esta asociación no se ha podido demostrar aún [8]. En 2 grandes cohortes, de 21 y 40 pacientes, no se identificaron variaciones somáticas ni de la línea germinal en el gen NLRP3 mediante secuenciación de nueva generación. Además, estos estudios tampoco detectaron ninguna otra variante patogénica en los paneles de secuenciación de nueva generación de genes conocidos por su implicación en enfermedades autoinflamatorias sistémicas [13,14]. No obstante, se encontró evidencia indirecta de la activación del inflamasoma, lo que se explica por los niveles elevados de agregados extracelulares de la proteína adaptadora tipo speck asociada a apoptosis en el suero de pacientes con SchS, que se liberan durante la piroptosis mediada por NLRP3 [13].
Aunque no se han encontrado alteraciones genéticas en los genes conocidos por su implicación en enfermedades autoinflamatorias, se ha descrito una variante somática en el gen MYD88, el cual codifica una proteína adaptadora implicada en las vías de señalización del receptor tipo Toll y del receptor de IL-1 [15]. Una mutación somática, con variación de un solo nucleótido en el exón 5 de MYD88, conduce a la sustitución de leucina por prolina en la posición 265 (L265P) [16]. L265P lleva a la supervivencia celular, al ensamblar espontáneamente un complejo proteico que contiene IRAK1 e IRAK4, lo que conduce a la actividad de la cinasa IRAK4, la fosforilación de IRAK1, la señalización de NF-κB, la activación de la vía JAK2/STAT3 y la secreción de IL-6, IL-10 e interferón-β en neoplasias malignas derivadas de células B [17]. En un estudio se evaluó la prevalencia de la mutación MYD88 L265P en 30 pacientes con SchS y 6 controles con CAPS y su estado de hematopoyesis oligoclonal. La variante MYD88 L265P se encontró en 9 de 30 pacientes con SchS, y se identificaron mutaciones somáticas asociadas con la hematopoyesis clonal en uno de 30 pacientes con SchS y uno de 6 pacientes con CAPS [15]. Otros autores también han reportado casos con identificación de la variante MYD88 L265P [18]. Con todos estos hallazgos, aún no es clara la asociación genética de esta entidad.
Finalmente, Wesselmann et al. describieron el caso de un paciente con SchS sin gammapatía monoclonal, en quien se empleó secuenciación de exoma completo y se encontraron variaciones en la subunidad B del factor de crecimiento derivado de plaquetas, la proteína tirosina cinasa TYRO3 y MALT1 (paracaspasa), genes que interactúan con la IL-1β que podrían tener un impacto en el fenotipo inflamatorio [19].
Rol de IL-1A pesar de que los mecanismos fisiopatológicos que subyacen en el SchS aún no están esclarecidos en su totalidad, muchos elementos sustentan que existe una activación del sistema inmunológico innato, puntualmente en el inflamosoma, y se resalta la IL-1β como un determinante central en la patogénesis de la enfermedad. Esto se ve demostrado en el control marcado de síntomas observado luego del tratamiento con antagonistas de IL-1 [20]. Sin embargo, al interrumpirse la inhibición de IL-1 hay un retorno de los síntomas, lo que evidencia que el proceso de la enfermedad sigue siendo impulsado por mecanismos que actúan antes de la liberación de IL-1β [21].
El papel de la IL-1β se demostró en un estudio que evaluó el efecto de varios ligandos del receptor tipo Toll sobre la producción de IL-1β, IL-6 y el factor de necrosis tumoral alfa (TNF-α) por células mononucleares de sangre periférica (PBMC) de 8 pacientes que cumplían criterios para SchS, incluidos 2 pacientes con mosaicismo NLRP3. In vitro, se encontró que la estimulación con LPS dio como resultado una mayor producción de IL-6 e IL-1β en PBMC de pacientes sintomáticos con SchS, en comparación con controles sanos [22]. Adicionalmente, varios estudios han demostrado que el SchS se caracteriza por mayores niveles circulantes de IL-18 libre, lo que posiblemente conduzca a una mayor activación de las células efectoras innatas/inflamatorias [23].
En una cohorte prospectiva se seleccionaron muestras de sangre de 36 pacientes con SchS tratados y no tratados con anakinra, un anti-IL-1. Se cultivaron PBMC con y sin LPS o anti-CD3/CD28 y se evaluaron los niveles de citocinas en suero. Se encontró una mayor liberación espontánea de TNF-α, IL-6, IL-1β, IL-1α y el antagonista del receptor de la IL-1 por parte de las PBMC de pacientes con SchS, en comparación con los controles. También se encontraron niveles reducidos de las citocinas Th1 (IFN-γ), Th2 (IL-4), Th17 (IL-17) y Treg (IL-10). Este hallazgo respalda la hipótesis de que la sobreproducción sistémica de IL-1β podría ocasionar una disfunción en las células T Th1, Th2, Th17 y Treg. Resulta de interés considerar que los niveles de citocinas tienden a normalizarse en pacientes tratados con anakinra. Esta observación demuestra además que la eficacia de la anakinra podría estar vinculada a la restauración de las funciones de las células Th, lo que amerita mayor profundización con otros estudios [24].
Por otro lado, se ha observado que los niveles de la quimiocina CCL2 están elevados en pacientes con SchS, mostrando una correlación directa con la actividad global de la enfermedad [11]. Sobre la propuesta del rol de CCL2 en la patogenia de la enfermedad, se plantea que la IL-1β induce la producción de esta quimiocina en hueso y piel, atrayendo monocitos a través del receptor CCR2. Estas células seleccionadas generan un bucle de amplificación patogénica al producir más IL-1β y CCL2. En el hueso, los monocitos seleccionados se diferencian en osteoclastos, bajo la influencia de IL-1β, TNF-α, RANKL y M-CSF, lo que da lugar a osteoesclerosis y dolor óseo. En la piel, los monocitos se convierten en macrófagos y células dendríticas, que producen IL-1β y TNF-α. Junto con la IL-17, esto induce la expresión de ligandos en los queratinocitos, lo que atrae neutrófilos y genera la dermatitis neutrofílica típica en este síndrome [25].
Disfunción de células Th17Diversos estudios han demostrado que las células Th17 pueden asumir funciones proinflamatorias o antiinflamatorias, dependiendo de su perfil de citocinas [26].
Las células Th17, que pueden coproducir IL-10, tienen funciones reguladoras similares a las de las células Treg, debido a su capacidad para suprimir la proliferación de células T helper, sin embargo, las células Th17, que se generan en presencia de IL-1β, no tienen estas propiedades [27,28].
En el estudio realizado por Noster et al., mediante ensayos de supresión, se evaluaron células Th17 dependientes e independientes de IL-1β en la modulación de la secreción de citocinas proinflamatorias antes y durante la terapia con anti IL-1β. Se demostró que en pacientes con SchS, la sobreproducción sistémica de IL-1β conduce a una pérdida profunda de las funciones antiinflamatorias de las células Th17, debido a la no supresión de la proliferación de células T naive. Es importante tener en cuenta que este fenómeno se revertía con el tratamiento anti-IL-1β [29].
Trampas extracelulares de neutrófilosLos neutrófilos activados liberan trampas extracelulares de neutrófilos (NET) en respuesta a una variedad de estímulos, en un proceso conocido como NETosis, que consiste en la liberación de gránulos intracelulares que capturan y destruyen microorganismos como virus, hongos, bacterias y protozoos [30].
La hipótesis principal sobre el papel de las NET en el SchS postula que los mastocitos hipersensibles a patrones moleculares asociados a daño o a los patrones moleculares asociados a patógenos se activan a través de los receptor tipo Toll y producen IL-1β y IL-6, presumiblemente mediados por el inflamasoma NLRP3. Estas citocinas proinflamatorias conducen a la selección de neutrófilos de la sangre periférica. Las citocinas y otros factores séricos inducen la formación inicial de NET en la piel. Durante la NETosis, se secretan IL-17 y LL-37 (catelicidina). La IL-17 induce una mayor selección de neutrófilos, mientras que LL-37 activa el inflamasoma NLRP3 en los macrófagos para producir más IL-1β, lo que a su vez promueve la selección adicional de estas células [31].
Bonnekoh et al. demostraron que, a diferencia de los pacientes con otras dermatosis neutrofílicas como síndrome de Sweet, CAPS, pioderma gangrenoso, vasculitis urticarial y urticaria crónica espontánea, así como controles sanos, los pacientes con SchS presentaron tasas de NETosis mucho más altas, y esto se correlacionó con niveles elevados de proteína C reactiva (PCR) y la presencia de NETosis, significativamente más visible en los pacientes con SchS [32]. Estos hallazgos refuerzan que los neutrófilos pueden tener un papel importante en la fisiopatología del SchS.
Rol de las células BUno de los datos más llamativos y que generan más inquietud en la enfermedad es el rol que desempeña la inmunoglobulina M (IgM) en la patogénesis. En los pacientes con SchS se ha observado gammapatía monoclonal IgM asociada con la cadena ligera kappa en la gran mayoría de los casos, o con menos frecuencia la inmunoglobulina G (IgG), lo cual evidencia que las células B podrían cumplir un papel dentro de la fisiopatología [33]. Mediante secuenciación de nueva generación de las regiones VDJ de los receptores de células B en 10 pacientes, se logró establecer que las células B corresponden a varias clonas y no reconocen un antígeno determinado, y aunque hay una expansión de estas células, esta es policlonal. Además, se intentó demostrar el rol de la paraproteína, considerando un posible reconocimiento de autoantígenos, sin embargo, esta posibilidad fue descartada [34].
El hecho de que la gammapatía monoclonal no se resuelva con el tratamiento anti-IL-1 es un dato que genera incertidumbre, y plantea la posibilidad de que IL-1 no es responsable de la expansión o el crecimiento clonal de las células B; sin embargo, pueden estar implicados otros factores [35], como lo demuestran estudios en los cuales la IL-18 participa en el crecimiento de las células B y la producción de anticuerpos, y, a su vez, las células B expresan receptores de IL-18 funcionales [36]. Llama la atención, además, que Bhattacharyya et al. demostraran que la IL-18 fue producida por células B monoclonales IgM+ en un paciente con SchS, y después del tratamiento con rituximab (un anticuerpo contra CD20) los niveles de IL-18 e IgM disminuyeron [37]. Todo lo anterior plantea como hipótesis de la patogénesis del SchS, la activación simultánea del inflamosoma por un desencadenante desconocido, que a su vez generaría la activación de células B, la cual también puede verse influida por la participación de IL-18.
Con respecto al papel de las células B en la enfermedad, es aún más retador encontrar la descripción de casos sin gammapatía monoclonal pero con las típicas lesiones urticariformes, adenopatías, fiebre y organomegalia; los autores de estos casos explican la ausencia de gammapatía, posiblemente como un hallazgo temporal, y plantean la posibilidad de que esta aparezca más tarde en la evolución [38].
En la figura 2 se esquematizan los eventos fisiopatológicos que subyacen en el SchS.
.")
Hipótesis de la patogénesis del síndrome de Schnitzler. Un estímulo desconocido activa NLRP3 y una célula B. La actividad de IL-1β media las manifestaciones infamatorias, mientras que la activación de la célula B por el estímulo de la IL-18 explica la gammapatía monoclonal. La IL-1β además participa en la selección de neutrófilos y en la diferenciación de células de Th17 con propiedades proinflamatorias. A su vez, la IL-17 aumenta la selección de neutrófilos.
Fuente: Creada con BioRender (elaboración propia).
La caracterización más completa conocida acerca de la presentación clínica y los hallazgos de laboratorio documentados en los pacientes con esta enfermedad se encuentra en el informe de De Koning, con 281 casos. La evidencia publicada hasta el momento describe como hallazgo distintivo de la enfermedad la urticaria crónica, asociada con gammapatía monoclonal, aunado esto a síntomas constitucionales como fiebre intermitente, artralgias, fatiga y dolor óseo como características relacionadas con mayor frecuencia [4]. Otras manifestaciones se han documentado en menor proporción, como el angioedema, la pérdida de peso, la hepatoesplenomegalia, la neuropatía y la sudoración nocturna [7]. Las manifestaciones clínicas, así como la descripción de su frecuencia en la literatura, se esquematizan en la figura 3. A continuación, se amplían las características semiológicas de los rasgos más representativos de la enfermedad.
.")
La urticaria, por definición, está presente en el 100% de los pacientes con SchS. Con mayor frecuencia, afecta el tronco y las extremidades, y suele respetar el rostro, el cuello, las palmas y las plantas. Por lo general, las erupciones persisten durante menos de 48h [6]. En la mayor parte de los pacientes, se observa como máculas rosadas a rojas o pápulas ligeramente elevadas o en forma de placas [39]. La frecuencia de las erupciones urticariales puede variar entre pacientes, desde una presentación diaria hasta erupciones cutáneas pocas veces al año [40]. El hallazgo histopatológico cutáneo más frecuente es la inflamación neutrofílica perivascular e intersticial, en un 57%, o inflamación perivascular de células predominantemente mononucleares, en un 29%, con eosinófilos en el 50% de los casos [4]. Adicionalmente, se ha documentado la presencia de depósitos de IgM en la dermis y en la epidermis [6].
FiebreDesde el punto de vista semiológico, se describe fiebre con patrón intermitente, de duración e intensidad variables, con valores incluso mayores de 40°C; puede asociarse a fatiga y sudoración nocturna. No se ha encontrado una correlación entre su aparición y el exantema [41]. Puede responder al tratamiento con antiinflamatorios no esteroideos o colchicina, sin embargo, la respuesta drástica al tratamiento con anti-IL-1 es uno de los datos característicos en esta enfermedad, similar al comportamiento de CAPS, como se ha mencionado [42].
Afección musculoesqueléticaEl compromiso musculoesquelético es otra de las manifestaciones más descritas de la enfermedad. El dolor óseo de predominio en el hueso pélvico y la tibia es la localización más común, pero también se puede presentar artralgia o artritis. Según una cohorte retrospectiva, la remodelación ósea anormal estuvo presente en el 64% de los pacientes con SchS [43]. Las lesiones óseas más comúnmente descritas son la osteoesclerosis focal, la hiperostosis, la reacción perióstica y el aumento de la captación de huesos largos en las gammagrafías óseas, que afecta con mayor frecuencia al fémur distal, la tibia proximal (signo conocido como «las rodillas calientes») y los huesos innominados [44].
La utilidad de la PET/TC con fluorodesoxiglucosa 18-F y la gammagrafía en el seguimiento de SchS se evaluó en un estudio, en el cual se encontró que la PET/TC no parece ser de mucha utilidad en el seguimiento de la enfermedad, mientras que la gammagrafía ósea es más sensible para el diagnóstico y la determinación de hallazgos que pueden correlacionarse con la actividad clínica [45]. Estos datos también fueron constatados en el estudio de Darrieutort-Laffite et al., en el que la puntuación gammagráfica se correlacionó con la actividad clínica (r=0,4, p<0,02) y el nivel de PCR (r=0,47, p<0,01); asimismo, hubo disminución en la puntuación gammagráfica en los pacientes que fueron tratados con corticosteroides o anti-IL-1, en contraste con los pacientes no tratados [46].
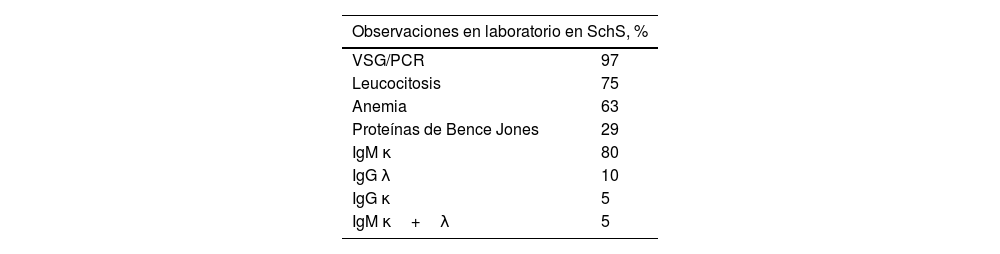
Hallazgos paraclínicosEn cuanto a los hallazgos de laboratorio, la característica más distintiva de la enfermedad es la gammapatía monoclonal. Cerca del 90% de los casos presenta gammapatía monoclonal de tipo IgM, mientras que el 10% restante se asocia en particular con gammapatía de tipo IgG, considerada una variante de la enfermedad [47]. Recientemente, también se han reportado algunos casos relacionados con gammapatía IgA [48]. Estas variantes de inmunoglobulinas amplían el espectro clínico de la enfermedad y plantean importantes desafíos diagnósticos. Además, se han reconocido casos de SchS sin gammapatía monoclonal, que suelen denominarse síndromes similares a Schnitzler[19].
Dada la relevancia de la gammapatía monoclonal en el diagnóstico de la enfermedad, es fundamental incluir en el enfoque diagnóstico la realización de electroforesis de proteínas séricas, inmunofijación y cuantificación de inmunoglobulinas.
Otras alteraciones comunes observadas en estos pacientes son leucocitosis, neutrofilia, anemia y elevación de reactantes de fase aguda como VSG y PCR (tabla 1) [7].
Hallazgos de laboratorio más comunes en el síndrome de Schnitzler
| Observaciones en laboratorio en SchS, % | |
|---|---|
| VSG/PCR | 97 |
| Leucocitosis | 75 |
| Anemia | 63 |
| Proteínas de Bence Jones | 29 |
| IgM κ | 80 |
| IgG λ | 10 |
| IgG κ | 5 |
| IgM κ+λ | 5 |
IgG: inmunoglobulina G; IgM: inmunoglobulina M; PCR: proteína C reactiva; SchS: síndrome de Schnitzler; VSG: velocidad de sedimentación globular.
Fuente: Adaptada de De Koning et al. [7].
Desde las primeras descripciones de la enfermedad, su definición ha evolucionado paulatinamente, sin embargo, en la actualidad aún no existe ningún marcador que pueda precisar el diagnóstico. Por lo tanto, el diagnóstico consiste en una conjugación de manifestaciones clínicas, biológicas e histológicas por medio de criterios que se han ido modificando a lo largo de los años [39]. Los primeros criterios propuestos fueron establecidos por Lipsker et al., quienes requerían como características esenciales urticaria y el componente de IgM monoclonal, además de al menos 2 de 8 características clínicas, de laboratorio e imagen [49]. Posteriormente, en el 2007, De Koning et al. modificaron estos criterios (tabla 2) e incluyeron la variante por gammapatía por IgG como uno de los criterios obligatorios, teniendo en cuenta que un pequeño porcentaje de estos pacientes puede presentar esta variante. Desde entonces, la gran mayoría de las publicaciones utilizan estos criterios.
Criterios modificados de Lipsker de síndrome de Schnitzler
| Erupción urticarial+IgM monoclonal (o IgG: tipo variante) y al menos 2 de los siguientes criterios: |
| FiebreArtralgia o artritisDolor de huesosGanglios linfáticos palpablesAgrandamiento del hígado o del bazoVelocidad de sedimentación elevadaLeucocitosisHallazgos anormales en las investigaciones morfológicas óseas |
Es necesario descartar otras causas en todos los casos, en particular el síndrome de hiper-IgD, la enfermedad de Still del adulto, la vasculitis urticarial hipocomplementémica, la deficiencia adquirida del inhibidor de C1 y la crioglobulinemia.
IgG: inmunoglobulina G; IgM: inmunoglobulina M.
Fuente: Adaptada de Lipsker et al. [49].
Estos criterios fueron revisados en 2013 por un grupo de expertos reunidos en Estrasburgo, quienes consideraron que era necesario distinguir entre pacientes con SchS definitivo y SchS probable, puesto que de esta manera se facilitarían los estudios patogénicos y terapéuticos. Estos criterios son conocidos como los «criterios de Estrasburgo» (tabla 3) [50].
Criterios de Estrasburgo
| Criterios diagnósticos de Estrasburgo | |
|---|---|
| Criterio obligatorio | Criterios menores |
| Erupción urticaria crónica+IgM monoclonal o IgG | Fiebre recurrenteaHallazgos objetivos de remodelación ósea anormal con o sin dolor óseobInfiltrado dérmico neutrofílico en la biopsia de pielcLeucocitosis y/o PCR elevadad |
| Diagnóstico definitivo → Dos criterios obligatorios y al menos 2 criterios menores si es IgM, y 3 criterios menores si IgG | |
| Diagnóstico probable → Dos criterios obligatorios y al menos un criterio menor si es IgM, y 2 criterios menores si es IgG | |
IgG: inmunoglobulina G; IgM: inmunoglobulina M.
Debe ser>38°C y no tener explicación alguna. Suele aparecer (pero no es obligatorio) junto con la erupción cutánea).
Dermatosis urticaria neutrofílica en ausencia de necrosis fibrinoide y edema dérmico significativo.
Leucocitosis (>10.000/mm3) y/o proteína C reactiva (PCR) elevada (>30mg/l).
Fuente: Adaptada de Simon et al. [50].
Es importante resaltar que antes de considerar el diagnóstico de SchS deben descartarse en todo paciente con lesiones cutáneas y fenotipo inflamatorio trastornos como la enfermedad de Still del adulto, los CAPS y otros síndromes autoinflamatorios monogénicos, vasculitis urticarial y crioglobulinémica, lupus eritematoso sistémico, urticaria idiopática crónica y gammapatía monoclonal de significado desconocido; estas 2 últimas afecciones, teniendo en cuenta que son relativamente comunes en la edad adulta y su coexistencia no es excepcional [1,50].
Gusdorf et al. llevaron a cabo un estudio multicéntrico entre 2009 y 2014, con el fin de evaluar la aplicabilidad y la validez de los criterios diagnósticos existentes en SchS, en el que se incluyeron 42 pacientes con SchS, 12 con enfermedad de Still de inicio en la edad adulta, 7 con CAPS, 9 con enfermedad de Waldenström y 10 con urticaria crónica espontánea. En este estudio todos los pacientes con SchS cumplieron los criterios de Lipsker; se determinó una sensibilidad y especificidad del 100 y el 97%, respectivamente. Con relación a los criterios de Estrasburgo, 34 pacientes tenían SchS definitivo, 5 tenían diagnóstico probable y 3 no cumplían los criterios. Para los criterios de Estrasburgo, la sensibilidad para el diagnóstico definitivo y probable fue del 81 y el 93%, respectivamente, con una especificidad correspondiente del 100 y el 97%. Por tanto, ambos criterios son confiables para el diagnóstico en la actualidad [51].
TratamientoLas terapias convencionales que se utilizan en el tratamiento de enfermedades como la urticaria, tales como antihistamínicos, antiinflamatorios, corticosteroides y fármacos inmunosupresores, suelen tener una eficacia moderada o ser ineficaces en el SchS. En la actualidad, la terapia estándar de oro son los agentes anti-IL-1, como anakinra, canakinumab o rilonacept, que han demostrado un control significativo de los síntomas clínicos [42]. Otras terapias, como los anti-TNF, los anti-IL-6 y los anti-CD20, se han usado en pacientes con respuestas variables. A continuación, se describen brevemente los resultados en la literatura para algunos de ellos.
Anti-IL-1βLa anakinra es un análogo estructural no glucosilado del antagonista de los receptores de interleucina que neutraliza la actividad biológica de la IL-1α e IL-1β, al inhibir competitivamente su unión al receptor tipo i de la IL-1. Se ha utilizado ampliamente en la enfermedad, con resultados beneficiosos en su gran mayoría [13,29,52]. En un estudio observacional se analizó retrospectivamente la eficacia y la seguridad a largo plazo de IL-1Rα y el resultado de los pacientes que no recibieron este tratamiento. Se encontró que en 29 pacientes que recibieron IL-1Rα hubo una respuesta clínica favorable. Después de una mediana de seguimiento de 36 meses (rango: 2-79), la eficacia se mantuvo sin cambios. Las fallas del tratamiento con este medicamento deben llevar a reconsiderar el diagnóstico [53].
El canakinumab es un anticuerpo monoclonal anti-IL-1β del isotipo IgG1/κ completamente humano [21]. Su eficacia fue evaluada en un ensayo clínico multicéntrico, aleatorizado y controlado con 20 pacientes con enfermedad activa, en comparación con placebo. En este estudio se encontró que los pacientes tratados con canakinumab tuvieron una respuesta clínica completa al séptimo día significativamente mayor (p=0,001), en comparación con el grupo de placebo. Asimismo, los niveles de los marcadores de inflamación, la PCR y el amiloide A sérico, y las puntuaciones de calidad de vida mejoraron de manera significativa en los individuos tratados con este medicamento [54].
El rinolacept es una proteína de fusión dimérica conformada por el dominio extracelular del receptor de la IL-1β y del antagonista del receptor de la IL-1β, fusionados con la porción Fc de la IgG. Esta proteína de fusión bloquea las acciones biológicas de las citocinas IL-1α e IL-1β [2]. Su eficacia se evaluó en un estudio prospectivo con 8 pacientes con la enfermedad mediante evolución clínica y marcadores séricos como PCR, amiloide A sérico y proteína A12 fijadora de calcio S100. Se encontró una respuesta clínica rápida, acompañada de reducciones en la PCR y el amiloide A sérico, que continuaron durante la duración del tratamiento [10].
Anti-IL-6El tocilizumab es un anticuerpo monoclonal humanizado recombinante de la subclase de las inmunoglobulinas IgG1, que se une específicamente a los receptores de IL-6 tanto solubles como unidos a la membrana (sIL-6R y mIL-6R), e inhibe la señalización de la IL-6 mediada a través de estos receptores [55]. En un ensayo clínico prospectivo abierto, se evaluó la eficacia terapéutica en 8 pacientes con SchS, sin embargo, a pesar de la respuesta inicial favorable, la mitad de los pacientes perdieron el beneficio después de 16 semanas y suspendieron el tratamiento, y luego de 52 semanas de observación, 3 de cada 4 pacientes también mostraron una recaída de los síntomas clínicos, a pesar de que la PCR permaneció normal [56]. En conclusión, el tratamiento con tocilizumab redujo los síntomas clínicos y los marcadores inflamatorios en el SchS. Debido a que con el paso del tiempo hay una pérdida de eficacia en la mayoría de los pacientes, su uso debe considerarse principalmente en personas que no responden a otras terapias dirigidas a citocinas [57].
Anti-CD20El rituximab es un anticuerpo monoclonal quimérico contra CD20 que se expresa en los linfocitos B. Este medicamento aún no se ha estudiado en ensayos clínicos, por lo cual los datos sobre el beneficio del bloqueo de células B en esta enfermedad proceden principalmente de reportes de casos [2]. Se ha observado que en alrededor del 20% de los casos mostró una respuesta significativa, mientras que un 16% tuvo una respuesta parcial al tratamiento con este medicamento. Estos casos de respuesta al rituximab abarcaron aquellos que estaban recibiendo una terapia combinada del medicamento junto con quimioterapia o radiación para enfermedades malignas [58].
Anti-TNFCon respecto a los inhibidores del TNF, solo el adalimumab mostró beneficio en un paciente. Otros, como el infliximab o el etanercept, no mostraron beneficio o exacerbaron los síntomas [2].
ConclusionesEl SchS es una enfermedad cuyas bases fisiopatológicas son en gran medida desconocidas. Dado que esta enfermedad puede imitar a otras como el lupus eritematoso sistémico, la enfermedad de Still del adulto o la vasculitis, debe considerarse como un diagnóstico de exclusión. Se debe sospechar en pacientes con dermatosis urticariales, fiebre de origen desconocido y artralgias. El reconocimiento temprano impacta en la calidad de vida de estos pacientes, teniendo en cuenta que la remisión espontánea de los síntomas es poco probable y hay un porcentaje considerable de casos que pueden progresar a enfermedad maligna. Aún se requieren estudios para ampliar la fisiopatología de la enfermedad y su pronóstico a largo plazo.
AutoríaTodos los autores participaron en la redacción del manuscrito y en su aprobación final.
FinanciaciónLos autores no recibieron financiación para realizar este artículo.
Consideraciones éticasEsta revisión se cataloga como un proyecto sin riesgo, puesto que no incluye pacientes y su contenido se basa solo en una revisión exhaustiva de la literatura.
Declaración sobre el uso de la IA generativa y de las tecnologías asistidas por la IA en el proceso de redacciónDurante la preparación de este trabajo los autores utilizaron ChatGPT 4.0 como apoyo en la traducción de artículos. Tras utilizar dicha herramienta, revisaron y editaron el contenido según la necesidad, y asumen la plena responsabilidad del contenido de la publicación.
Ninguno.
Al doctor Estiven Crespo por la revisión de la redacción del artículo.
