

















Las ictiosis hereditarias son un grupo de trastornos genéticos de la cornificación, que se caracterizan por presentar hiperqueratosis y/o descamación. La nueva clasificación identifica 36 tipos de ictiosis, las cuales se subdividen según su frecuencia, patrón de herencia y compromiso extracutáneo. El diagnóstico se basa principalmente en las características clínicas, ya que los estudios genéticos no se encuentran disponibles en nuestro medio. El tratamiento es sintomático y su manejo debe ser realizado por un equipo multidisciplinario. En este artículo se revisan los aspectos diagnósticos y terapéuticos de los distintos tipos de ictiosis, considerando la nomenclatura y modificaciones expuestas en la nueva clasificación.
Hereditary ichthyoses are a group of genetic disorders of cornification, which are characterised by hyperkeratosis and scaling. The new classification identifies 36 types of ichthyosis, which are subdivided according to their frequency, pattern of inheritance and extracutaneous involvement. The diagnosis is mainly based on clinical features, since genetic studies are not available in our setting. Treatment is symptomatic and management should be performed by a multidisciplinary team. In this article, the diagnostic and therapeutic aspects of different types of ichthyosis are reviewed, taking into account the nomenclature and modifications presented in the new classification.
El término ictiosis define un grupo de trastornos generalizados de la cornificación, que se caracterizan por presentar hiperqueratosis y/o descamación1. Su nombre deriva de la palabra griega icthys que significa pez, debido al aspecto escamoso de la piel afectada2.
Las ictiosis hereditarias corresponden a trastornos genéticos de la cornificación, a diferencia de las ictiosis adquiridas, que pueden ser secundarias a neoplasias malignas, enfermedades autoinmunes, inflamatorias, infecciosas, metabólicas, reacciones medicamentosas y deficiencias nutricionales3.
En el año 2009 un grupo de expertos desarrolló una nueva clasificación de consenso basada principalmente en las características clínicas, pero considerando los aspectos fisiopatológicos y moleculares descubiertos hasta el momento, lo que ha permitido facilitar la comprensión de la enfermedad y el estudio de los pacientes. Esta clasificación identifica 36 tipos de ictiosis, las cuales se dividen en subgrupos de acuerdo a la presencia o no de compromiso extracutáneo, frecuencia de la enfermedad y patrón de herencia1 (tabla 1).
Clasificación clínica según el consenso de ictiosis 2009
| Ictiosis no sindrómicas | Ictiosis sindrómicas |
|---|---|
| Ictiosis comunes | Ligadas a X |
| Ictiosis vulgar Ictiosis recesiva ligada a X | Ictiosis recesiva ligada a X Síndrome IFAP Síndrome de Conradi-Hunermann-Happle |
| ICAR | Autosómicas |
| Formas mayores Ictiosis lamelar Eritrodermia ictiosiforme congénita Ictiosis arlequín | Trastornos del pelo Síndrome de Netherton Tricotiodistrofia Síndrome IH Síndrome IHCE |
| Formas menores Bebe colodión autorresolutivo Bebe colodión autorresolutivo acral Ictiosis en traje de baño | |
| Trastornos neurológicos Síndrome de Sjogren Larsson Síndrome de Refsum Síndrome de MEDNIK | |
| Ictiosis queratinopáticas | |
| Formas mayores Ictiosis epidermolítica Ictiosis epidermolítica superficial | |
| Curso letal Síndrome de Gaucher tipo 2 Déficit múltiple de sulfatasas Síndrome de CEDNIK Síndrome ARC | |
| Formas menores Ictiosis epidermolítica autosómica recesiva Ictiosis epidermolítica anular Ictiosis Curth-Macklin Nevus epidermolítico | |
| Otros signos asociados Síndrome Chanarin-Dorfman Síndrome KID Síndrome ictiosis-prematuridad | |
| Otras formas | |
| Queratodermia loricrina Eritroqueratodermia variabilis Síndrome de piel exfoliada Eritrodermia ictiosiforme congénita reticular Síndrome KLICK |
ICAR: ictiosis congénitas autosómicas recesivas; IH: ictiosis-hipotricosis; IHCE: ictiosis-hipotricosis-colangitis esclerosante.
Fuente: Oji et al.1.
Las manifestaciones extracutáneas permiten identificar 2 grandes grupos de ictiosis. Cuando el defecto genético se manifiesta exclusivamente en la piel se denominan ictiosis no sindrómicas, mientras que la presencia de alteraciones en otros órganos o sistemas además de la piel, define las ictiosis sindrómicas1,3.
A continuación se describen las características de los diferentes tipos de ictiosis (tablas 2 y 3), con énfasis en los tipos más clásicos.
Características de ictiosis no sindrómicas
| Tipo de ictiosis | Gen afectado | Herencia | Características clínicas |
|---|---|---|---|
| Ictiosis vulgar | FLG | AD | Se inicia en la infancia. Se caracteriza por xerosis, descamación generalizada color blanquecino, sin eritema basal. Respeta las fosas poplíteas y antecubitales. Se asocia a hiperlinearidad palmoplantar |
| Ictiosis recesiva ligada a X | STS | XR | Se presenta al nacimiento con eritrodermia y/o descamación generalizada. Se caracteriza por presentar escamas grandes, oscuras, adheridas, generalizadas, sin eritema. Respeta la cara, los pliegues, las palmas y las plantas |
| Ictiosis lamelar | TGM1/ALOXE3/ ALOX12B/NIPAL4/ CYP4F22/ ABCA12, otros | AR | Se presenta al nacimiento como bebé colodión. Se caracteriza por presentar escamas grandes, oscuras, generalizadas. No respeta pliegues Se asocia a ectropión, alopecia cicatricial, alteraciones ungueales e hipohidrosis |
| Eritrodermia ictiosiforme congénita | ALOXE3/ALOX12B, TGM1/NIPAL4/CYP4F22/ABCA12, otros | AR | Se presenta al nacimiento como bebé colodión. Se caracteriza por presentar escamas blancas, finas, generalizadas, con eritema de base Se puede asociar a ectropión leve, alopecia cicatricial, alteraciones ungueales e hipohidrosis |
| Ictiosis arlequín | ABCA12 | AR | Al nacimiento se presenta como bebe colodión severo. La presentación clínica es similar a un cuadro severo de IL o EIC |
| Bebe colodión autorresolutivo | TGM1, ALOX12B, ALOXE3 | AR | Se presenta al nacimiento como bebe colodión, pero se resuelve en forma completa o casi completa a los 3 meses |
| Bebe colodión autorresolutivo acral | TGM1 | AR | Al nacimiento presenta membrana colodión acral, la cual se resuelve en forma completa a los 3 meses |
| Ictiosis en traje de baño | TGM1 | AR | Se presenta al nacimiento. Se caracteriza por descamación tipo IL en el tronco, las axilas y el cuero cabelludo (áreas de mayor temperatura) |
| Ictiosis epidermolítica | KRT1/KRT10 | AD | Se presenta al nacimiento con eritrodermia, descamación leve y grandes erosiones. Se caracteriza por presentar hiperqueratosis con patrón en empedrado, más prominente en articulaciones y zonas de fricción. Se asocia a mal olor, eritema, ampollas tras traumatismos, infecciones cutáneas recurrentes y prurito. Queratodermia palmoplantar en caso de mutación en KRT1 |
| Ictiosis epidermolítica superficial | KRT2 | AD | Al nacimiento presenta eritrodermia y ampollas. Se caracteriza por hiperqueratosis sobre articulaciones, áreas denudadas superficiales, ampollas tras traumatismos, prurito sin eritema y sin afectación palmoplantar |
| Ictiosis epidermolítica autosómica recesiva | KRT10 | AR | Ídem a la forma autosómica dominante |
| Ictiosis epidermolítica anular | KRT1/KRT10 | AD | Se inicia dentro del primer año. Se caracteriza por descamación generalizada, ampollas y placas eritemato-descamativas, anulares o policíclicas en el tronco y las extremidades, de curso intermitente |
| Ictiosis de Curth-Macklin | KRT1 | AD | Se inicia en la infancia. Se caracteriza por presentar queratodermia palmoplatar difusa o estriada y placas hiperqueratósicas sobre las articulaciones, el tronco y las extremidades. Puede cursar con o sin eritrodermia |
| Nevus epidermolítico | KRT1/KRT 10 | Mosaico | Se presenta al nacimiento o en la infancia. Corresponde a una lesión hiperqueratósica, marronácea de disposición lineal que sigue las líneas de Blaschko |
| Queratodermia loricrina | LOR | AD | Se presenta al nacimiento como bebé colodión. Se caracteriza por presentar queratodermia palmoplantar difusa en panal de abeja, constricciones digitales y descamación fina generalizada con hiperqueratosis en articulaciones |
| Eritroqueratodermia variabilis | GJB3/GJB4 | AD | Se presenta al nacimiento o en el primer año de vida. Se caracteriza por placas eritematosas migratorias transitorias y placas hiperqueratósicas generalizadas o de límites geográficos de curso intermitente. Se asocia a queratodermia palmoplantar difusa |
| Síndrome de la piel exfoliada | -- | AR | Al nacimiento o en las primeras semanas de vida. Se caracteriza por presentar descamación exfoliativa, blanquecina, generalizada asociada a eritema, prurito y afectación palmoplantar. Curso intermitente |
AD: autosómica dominante, AR: autosómica recesiva, XR: recesiva ligada a X.
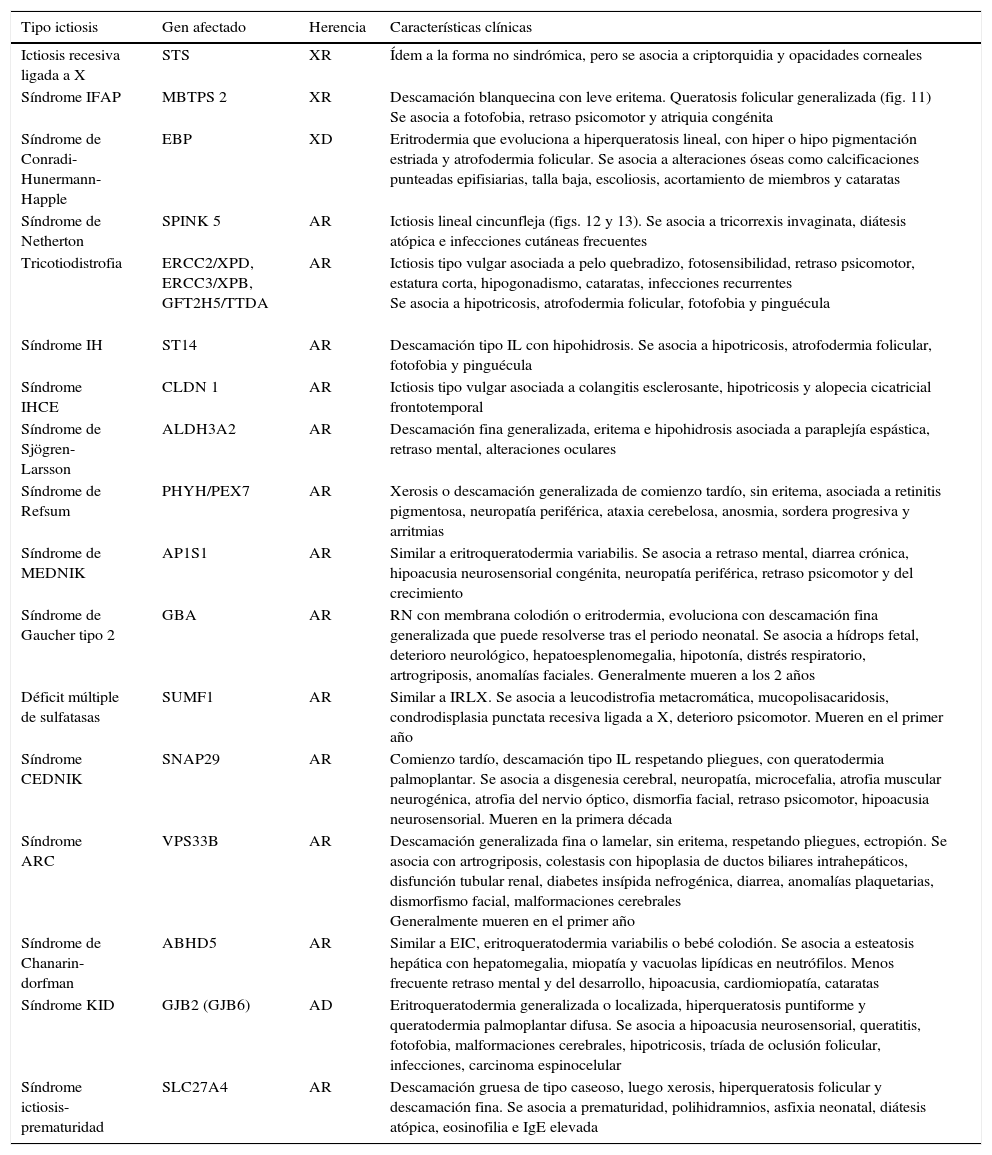
Características clínicas de ictiosis sindrómicas
| Tipo ictiosis | Gen afectado | Herencia | Características clínicas |
|---|---|---|---|
| Ictiosis recesiva ligada a X | STS | XR | Ídem a la forma no sindrómica, pero se asocia a criptorquidia y opacidades corneales |
| Síndrome IFAP | MBTPS 2 | XR | Descamación blanquecina con leve eritema. Queratosis folicular generalizada (fig. 11) Se asocia a fotofobia, retraso psicomotor y atriquia congénita |
| Síndrome de Conradi-Hunermann-Happle | EBP | XD | Eritrodermia que evoluciona a hiperqueratosis lineal, con hiper o hipo pigmentación estriada y atrofodermia folicular. Se asocia a alteraciones óseas como calcificaciones punteadas epifisiarias, talla baja, escoliosis, acortamiento de miembros y cataratas |
| Síndrome de Netherton | SPINK 5 | AR | Ictiosis lineal cincunfleja (figs. 12 y 13). Se asocia a tricorrexis invaginata, diátesis atópica e infecciones cutáneas frecuentes |
| Tricotiodistrofia | ERCC2/XPD, ERCC3/XPB, GFT2H5/TTDA | AR | Ictiosis tipo vulgar asociada a pelo quebradizo, fotosensibilidad, retraso psicomotor, estatura corta, hipogonadismo, cataratas, infecciones recurrentes Se asocia a hipotricosis, atrofodermia folicular, fotofobia y pinguécula |
| Síndrome IH | ST14 | AR | Descamación tipo IL con hipohidrosis. Se asocia a hipotricosis, atrofodermia folicular, fotofobia y pinguécula |
| Síndrome IHCE | CLDN 1 | AR | Ictiosis tipo vulgar asociada a colangitis esclerosante, hipotricosis y alopecia cicatricial frontotemporal |
| Síndrome de Sjögren-Larsson | ALDH3A2 | AR | Descamación fina generalizada, eritema e hipohidrosis asociada a paraplejía espástica, retraso mental, alteraciones oculares |
| Síndrome de Refsum | PHYH/PEX7 | AR | Xerosis o descamación generalizada de comienzo tardío, sin eritema, asociada a retinitis pigmentosa, neuropatía periférica, ataxia cerebelosa, anosmia, sordera progresiva y arritmias |
| Síndrome de MEDNIK | AP1S1 | AR | Similar a eritroqueratodermia variabilis. Se asocia a retraso mental, diarrea crónica, hipoacusia neurosensorial congénita, neuropatía periférica, retraso psicomotor y del crecimiento |
| Síndrome de Gaucher tipo 2 | GBA | AR | RN con membrana colodión o eritrodermia, evoluciona con descamación fina generalizada que puede resolverse tras el periodo neonatal. Se asocia a hídrops fetal, deterioro neurológico, hepatoesplenomegalia, hipotonía, distrés respiratorio, artrogriposis, anomalías faciales. Generalmente mueren a los 2 años |
| Déficit múltiple de sulfatasas | SUMF1 | AR | Similar a IRLX. Se asocia a leucodistrofia metacromática, mucopolisacaridosis, condrodisplasia punctata recesiva ligada a X, deterioro psicomotor. Mueren en el primer año |
| Síndrome CEDNIK | SNAP29 | AR | Comienzo tardío, descamación tipo IL respetando pliegues, con queratodermia palmoplantar. Se asocia a disgenesia cerebral, neuropatía, microcefalia, atrofia muscular neurogénica, atrofia del nervio óptico, dismorfia facial, retraso psicomotor, hipoacusia neurosensorial. Mueren en la primera década |
| Síndrome ARC | VPS33B | AR | Descamación generalizada fina o lamelar, sin eritema, respetando pliegues, ectropión. Se asocia con artrogriposis, colestasis con hipoplasia de ductos biliares intrahepáticos, disfunción tubular renal, diabetes insípida nefrogénica, diarrea, anomalías plaquetarias, dismorfismo facial, malformaciones cerebrales Generalmente mueren en el primer año |
| Síndrome de Chanarin-dorfman | ABHD5 | AR | Similar a EIC, eritroqueratodermia variabilis o bebé colodión. Se asocia a esteatosis hepática con hepatomegalia, miopatía y vacuolas lipídicas en neutrófilos. Menos frecuente retraso mental y del desarrollo, hipoacusia, cardiomiopatía, cataratas |
| Síndrome KID | GJB2 (GJB6) | AD | Eritroqueratodermia generalizada o localizada, hiperqueratosis puntiforme y queratodermia palmoplantar difusa. Se asocia a hipoacusia neurosensorial, queratitis, fotofobia, malformaciones cerebrales, hipotricosis, tríada de oclusión folicular, infecciones, carcinoma espinocelular |
| Síndrome ictiosis-prematuridad | SLC27A4 | AR | Descamación gruesa de tipo caseoso, luego xerosis, hiperqueratosis folicular y descamación fina. Se asocia a prematuridad, polihidramnios, asfixia neonatal, diátesis atópica, eosinofilia e IgE elevada |
AD: autosómica dominante, AR: autosómica recesiva; ARC: artrogriposis-disfunción renal-colestasis; IFAP: ictiosis folicular-atriquia-fotofobia; IH: ictiosis-hipotricosis; IHCE: ictiosis-hipotricosis-colangitis esclerosante; RN: recién nacido; XD: dominante ligada al cromosoma X; XR: recesiva ligada al cromosoma X.
Es la forma más común de ictiosis, con una prevalencia de 1:250-1.000 habitantes3. Presenta un patrón de herencia autosómica dominante y penetrancia incompleta2.
Se produce por mutaciones inactivantes del gen que codifica la filagrina. El déficit de filagrina altera la diferenciación de los corneocitos y la formación del factor natural de humectación, generando hiperqueratosis por retención y mayor pérdida transepidérmica de agua (TEWL)2,3.
En general, se inicia en la infancia con xerosis, descamación, prurito y eritema. La descamación es fina, blanca grisácea, generalizada, con escamas más prominentes en la superficie extensora de las extremidades (principalmente cara pretibial) y tronco. Respeta pliegues y áreas de flexión como fosas poplíteas y antecubitales (fig. 1). La descamación en el cuero cabelludo, la frente y las mejillas es común en la infancia, pero disminuye en la pubertad. En las palmas y en las plantas se observa hiperlinearidad (fig. 2) y con frecuencia se observa queratosis pilar en brazos, muslos y piernas2,4.


Las manifestaciones cutáneas suelen empeorar en ambientes fríos o secos y mejoran con la edad.
Se asocia a dermatitis atópica, asma y rinitis alérgica, ya que la alteración de la barrera cutánea predispone a una mayor sensibilización alérgica3.
El estudio histopatológico muestra hiperqueratosis asociada a hipo o agranulosis. En la microcopia electrónica se pueden observar gránulos de queratohialina ausentes, escasos y/o anormales2,3.
El diagnóstico diferencial se realiza con xerosis asociada a diátesis atópica, otras ictiosis hereditarias e ictiosis adquiridas. Es importante considerar este diagnóstico en pacientes con dermatitis atópica que cursan con escamas de gran tamaño en las caras extensoras de las extremidades4.
Ictiosis recesiva ligada a XSegunda forma de ictiosis en frecuencia, con una prevalencia de 1:2.000-6.0003. Se produce por una deleción (90%) o mutación inactivante en el gen que codifica la enzima sulfatasa esteroidea localizado en el cromosoma X, por lo cual afecta a los hijos de madres portadoras2.
El déficit enzimático en la epidermis resulta en un aumento de sulfato de colesterol que inhibe la descamación y genera hiperqueratosis por retención. En la placenta de fetos afectados resulta en disminución de los niveles de estrógenos, afectando el inicio y progresión del trabajo de parto2,3.
Se presenta en el periodo neonatal con eritrodermia y/o descamación generalizada, evolucionando con escamas grandes, romboidales, oscuras, muy adherentes y generalizadas que respetan la cara, los pliegues, las palmas y las plantas (fig. 3). La afectación es simétrica y predomina en la región lateral y posterior del cuello con un aspecto de «cuello sucio» (fig. 4), región retroauricular y zonas extensoras2,4. No se observa compromiso ungueal, dental o mucoso. Tiende a mejorar en ambientes cálidos y húmedos, pero no disminuye con la edad.

.")
El estudio histopatológico muestra hiperqueratosis con estrato granuloso normal o hipergranulosis. En la microscopia electrónica se observan gránulos de queratohialina aumentados en número y tamaño2,3.
El déficit enzimático se puede objetivar a través de otros estudios como electroforesis de lipoproteínas y medición de actividad de sulfatasa esteroidea en leucocitos2,3.
El diagnóstico diferencial se realiza principalmente con ictiosis vulgar e ictiosis lamelar.
Ictiosis congénitas autosómicas recesivasCorresponden a trastornos infrecuentes de la cornificación, con una prevalencia estimada de 1:138.000-1:300.0003.
El termino bebé colodión hace referencia a un neonato, generalmente de pretérmino, que nace recubierto por una membrana gruesa, tensa y brillante, asociado a ectropión, eclabium e hipoplasia de cartílagos auriculares y nasales2,4 (fig. 5). La membrana colodión se desprende entre la primera y cuarta semana de vida, evidenciando el fenotipo definitivo5. El 60% de los casos corresponde a la manifestación inicial de eritrodermia ictiosiforme congénita o ictiosis lamelar4, sin embargo un 10% tendrá una piel normal después de la resolución de la membrana colodión (bebe colodión autorresolutivo). Otros cuadros que pueden presentarse como bebe colodión son el síndrome de Sjögren-Larsson, tricotiodistrofia, síndrome de Netherton y displasias ectodérmicas, entre otros5.
Ictiosis lamelar
Ictiosis severa, de herencia autosómica recesiva. La mayoría de los casos son producto de mutaciones en el gen que codifica la enzima transglutaminasa-1 (TGM-1), sin embargo se ha asociado a mutaciones en otros genes como ALOXE3, ALOX12B, NIPAL4, CYP4F22, ABCA12 y otros aún no identificados5.
La disminución o pérdida de función de la enzima TGM-1 altera la formación de la envoltura cornificada y la unión de los lípidos intercelulares a esta envoltura, resultando en un trastorno importante de la diferenciación y descamación, generando hiperqueratosis por retención2,3.
Se presenta al nacimiento principalmente como bebe colodión, evolucionando con escamas grandes, oscuras y gruesas, de distribución generalizada y patrón en mosaico, con mínima eritrodermia o sin ella, comprometiendo las superficies flexoras, la cara y el tronco (fig. 6).

Suele presentar ectropión, eclabión, hipoplasia de cartílagos nasales y auriculares, hipohidrosis con intolerancia al calor, alopecia cicatricial, alteraciones ungueales (hiperqueratosis subungueal, surcos, pits, paroniquia) y queratodermia palmo-plantar de intensidad variable2,4.
La histopatología muestra hiperqueratosis ortoqueratósica masiva y acantosis, en ocasiones con patrón psoriasiforme. La tasa de proliferación celular es normal o está ligeramente elevada5.
El diagnóstico diferencial se realiza con ictiosis ligada a X, eritrodermia ictiosiforme congénita y síndrome de Netherton.
Eritrodermia ictiosiforme congénitaForma menos severa que la IL, de herencia autosómica recesiva. Los genes afectados más frecuentes son ALOXE3 y ALOX12B, que codifican las lipooxigenasas epidérmicas, y al igual que en la IL se puede asociar a mutaciones en otros genes (NIPAL4, CYP4F22, TGM-1, ABCA12)5.
La disminución o ausencia de lipooxigenasas genera cuerpos lamelares defectuosos con acumulación de lípidos intracelulares, lo cual inhibe la diferenciación y estimula la proliferación de corneocitos, generando una hiperqueratosis por aumento de recambio2,3.
La mayoría se presenta al nacimiento como bebe colodión, que posteriormente desarrolla eritrodermia y descamación generalizada. Presenta escamas finas en la cara, el cuero cabelludo, el tronco y escamas «tipo plato» (adheridas en el centro y deprendidas en los bordes) en la superficie extensora de las piernas2,4 (fig. 7).

Los pacientes con mutaciones en ALOX12B muestran una descamación más discreta y blanquecina en comparación con los portadores de mutaciones en ALOXE3, que presentan escamas marronáceas y adherentes. La presencia de eritema, hiperqueratosis palmoplantar y acentuación de pliegues palmoplantares se asocia a defectos en el gen ALOX12B5.
Suelen presentar ectropión leve, alopecia cicatricial, alteraciones ungueales e intolerancia al calor por hipohidrosis4.
En la histopatología se observa una hiperqueratosis menos marcada, con paraqueratosis focal o extensa e hipergranulosis. En la microscopia electrónica se observan cuerpos lamelares alterados y en mayor número2,3.
El diagnóstico diferencial se realiza con ictiosis vulgar e ictiosis lamelar.
Ictiosis arlequínTipo de ictiosis grave, generalmente fatal, de herencia autosómica recesiva. Se produce por la mutación del gen ABCA12 que codifica para una proteína trasportadora de membrana. Defectos en este gen alteran el transporte de lípidos citoplasmáticos a los cuerpos lamelares y el transporte de proteasas, lo cual determina una disminución de los lípidos intercelulares que conduce a una hiperproliferación compensatoria, asociado a una alteración en la descamación2,3.
Se presenta al nacimiento como un bebe colodión severo, exhibiendo placas hiperqueratósicas extensas, separadas por fisuras profundas, que tienden a configurar patrones geométricos (como la vestimenta de un payaso arlequín). La marcada tensión de la piel genera ectropión, eclabium, hipoplasia de cartílagos auriculares y nasales, restricción ventilatoria, contracturas en flexión de las extremidades y bandas constrictivas en los dígitos u ortejos4,5. No suelen tener pestañas ni cejas, aunque el cuero cabelludo puede estar conservado. Además, pueden presentar hipoplasia o ausencia de uñas (fig. 8).

El diagnóstico diferencial se realiza con ictiosis lamelar grave.
Ictiosis queratinopáticasIctiosis epidermolítica (ex hiperqueratosis epidermolítica, eritrodermia ictiosiforme congénita ampollar o ictiosis bulosa)Ictiosis de herencia autosómica dominante. Se produce por mutaciones en los genes de queratina 1 y 10, ubicadas en el estrato espinoso. Las queratinas defectuosas forman filamentos intermedios alterados que conducen al colapso del citoesqueleto celular y, con ello, a la formación de ampollas. Al mismo tiempo, el defecto de la barrera cutánea estimula la proliferación de queratinocitos, resultando en una hiperqueratosis por aumento de recambio2,3.
Se presenta al nacimiento con eritrodermia severa, descamación leve y bulas o grandes erosiones. Durante la primera semana de vida se observan brotes de ampollas flácidas, de contenido seroso, distribuidas principalmente en el tronco y en las extremidades, que evolucionan a extensas áreas denudadas, húmedas, dolorosas y de mal olor (fig. 9). Posteriormente, evoluciona con hiperqueratosis verrucosa en empedrado o puntiaguda como las púas de un puercoespín (tipo hystrix), principalmente en articulaciones y pliegues, donde es frecuente el mal olor por colonización bacteriana, con formación de ampollas posterior a traumatismos2–4 (fig. 10).


La presencia de queratodermia palmoplantar se relaciona con mutaciones en el gen KTR1.

La histología muestra hiperqueratosis extensa asociada a lisis de queratinocitos suprabasales y ampollas («hiperqueratosis epidermolítica»)3.
El diagnóstico diferencial en recién nacidos se establece con epidermólisis bulosa, síndrome de piel escaldada estafilocócica, necrólisis epidérmica tóxica y otras enfermedades ampollares2.
Ictiosis epidermolítica superficial (ictiosis bulosa de Siemmens)

Forma menos severa que IE. Se produce por mutación del gen KTR2, presente el estrato espinoso superior y granuloso, generando ampollas más superficiales y menor hiperqueratosis2,3.
Se presentan desde el nacimiento con eritrodermia y ampollas, evolucionando con hiperqueratosis y ampollas luego de traumatismos, que dejan áreas denundadas en «collarete»2.
DiagnósticoEs importante realizar una historia clínica detallada y examen físico completo con énfasis en el fenotipo cutáneo (patrón de descamación e hiperqueratosis, calidad y color de las escamas), inicio y evolución (bebe colodión, eritrodermia al nacimiento o ictiosis de inicio tardío), presencia de otras características dermatológicas (eritema, prurito, ampollas, erosiones, alteración de fanéreos), compromiso extracutáneo e historia familiar (patrón de herencia)2,6.
Estudio histopatológicoRequiere una cuidadosa correlación con los hallazgos clínicos y en algunos casos aporta información relevante para el diagnóstico. En el caso de ictiosis vulgar, la reducción o ausencia del estrato granuloso es característico. En la ictiosis epidermolítica, la presencia de ampollas microscópicas y fisuras acantolíticas orientan al diagnóstico, aun cuando no se observen erosiones o ampollas1,6.
La observación de pelos a la microscopia también aporta datos interesantes. La alteración en tallo de bambú o tricorrexis invaginata es un criterio diagnóstico de síndrome de Netherton. En cambio, la visualización de pelos atigrados con luz polarizada es característica de tricotiodistrofia6.
La microscopia electrónica es otra herramienta muy útil, aunque poco accesible en nuestro medio. La disminución de gránulos de queratohialina es común en la ictiosis vulgar. La disrupción del citoesqueleto, separación de desmosomas y secreción anormal de cuerpos lamelares se observa en las ictiosis queratinopáticas1,6.
Test genéticosActualmente se han identificado más de 50 mutaciones genéticas asociadas, lo cual permite realizar un diagnóstico más preciso y un adecuado consejo genético a las familias afectadas1,6; sin embargo, no se encuentra disponible en Chile.
TratamientoEl tratamiento es sintomático y debe ser individualizado, ya que la efectividad y tolerancia de cada paciente es diferente. Es importante considerar la edad, el tipo y gravedad de la ictiosis, la extensión y/o localización de las lesiones y la respuesta a terapias previas5,6.
Es fundamental educar tanto al paciente como a su familia respecto al carácter crónico de esta enfermedad y la importancia de un tratamiento permanente, que si bien no es curativo, permite aliviar los síntomas y mejorar la calidad de vida.
El dermatólogo tiene un rol fundamental en el diagnóstico, tratamiento y seguimiento, sin embargo, el manejo debe ser realizado por un equipo multidisciplinario7.
Medidas generalesEs recomendable que los pacientes se bañen a diario para eliminar las escamas, los residuos de crema y disminuir la carga bacteriana5. En general, se recomiendan baños cortos utilizando sustitutos de jabón (syndet), seguido de la aplicación inmediata de emolientes. En pacientes con escamas gruesas, los baños de inmersión y la remoción mecánica de escamas son especialmente útiles. El uso de jabones antisépticos o baños con dosis bajas de hipoclorito ayudan a disminuir la carga bacteriana, por lo que se recomiendan en caso de infecciones recurrentes y mal olor, el cual es secundario a la digestión de escamas por la microbiota cutánea5,6.
Tratamiento tópicoLos emolientes y queratolíticos tópicos suelen ser la primera línea de tratamiento, ya que mejoran la función de barrera y facilitan la descamación al ser aplicados al menos 2 veces al día7.
Para hidratar la piel se utilizan preparados con urea, glicerol o vaselina. En pacientes con escamas gruesas e hiperqueratosis marcada se puede añadir uno o más agentes queratolíticos como urea en altas concentraciones, ácido láctico, ácido salicílico y propilenglicol5,7.
La oclusión aumenta la efectividad de agentes emolientes o queratolíticos, por lo que se recomienda en áreas circunscritas y resistentes al tratamiento por periodos limitados5.
Los moduladores de la diferenciación de queratinocitos, como retinoides tópicos (tretinoína, adapaleno y tazaroteno) y derivados de vitamina D (calcipotriol), son especialmente útiles en áreas hiperqueratósicas, pero pueden causar irritación local5,7. Los preparados con N-acetilcisteína al 10% en una base water in oil (w/o), han demostrado ser efectivos, seguros y bien tolerados en pacientes con IL, mostrando una notable respuesta a las semanas de uso8,9. Del mismo modo, se han reportado buenos resultados con el uso combinado de colesterol 2% y lovastatina 2% loción en pacientes con síndrome de CHILD desde las 4-6 semanas de tratamiento10.
En los neonatos y lactantes, se recomienda utilizar un vehículo sin medicación, dado que la piel es más sensible, menos tolerante a los queratolíticos y presenta un mayor riesgo de absorción percutánea de productos tópicos como urea y ácido salicílico, entre otros5,11.
Respecto a la selección de tratamiento, es importante considerar que las ICAR responden bien al uso de queratolíticos potentes, mientras que las ictiosis queratinopáticas tienden a empeorar con su uso.
Tratamiento sistémicoLos retinoides orales (isotretinoína y acitretin) son derivados de la vitamina A que actúan regulando la proliferación y diferenciación celular, previniendo la hiperqueratosis y facilitando la descamación. En general, se reservan para ictiosis graves o refractarias al tratamiento tópico5,12. Se recomienda iniciar con dosis bajas, titulando de acuerdo a la respuesta del paciente hasta alcanzar la dosis mínima efectiva y mantener un seguimiento estrecho por sus importantes efectos adversos (alteraciones mucocutáneas, teratogenicidad, alteraciones musculoesqueléticas, metabolismo lipídico y hepático). Una alternativa a los retinoides sistémicos son los bloqueadores del metabolismo del ácido retinoico como liarozole, el cual ha demostrado una eficacia equivalente a los retinoides orales, con un mejor perfil de tolerancia12.
Cuidados especialesEn caso de ectropión, el uso de lágrimas artificiales, lubricantes oculares y la hidratación del rostro disminuyen la xeroftalmia y la retracción palpebral, por lo que la cirugía se reserva para casos de difícil manejo5,13. La limpieza periódica del conducto auditivo por un otorrinolaringólogo evita la acumulación de escamas previniendo la hipoacusia5.
Respecto a la actividad física, se recomienda evitar ejercicios extenuantes cuando la temperatura es alta, ya que la hipohidrosis aumenta el riesgo de golpes de calor y convulsiones5. La indicación de terapia física controlada es importante para evitar contracturas en flexión, especialmente en pacientes con IA. En caso de isquemia secundaria a bandas constrictivas, se recomienda tratamiento quirúrgico13.
En relación con el ámbito psicológico, la apariencia anormal de la piel puede generar problemas en la autoestima y adaptación social, por lo tanto, es muy importante el apoyo familiar y de los servicios de salud. Los padres deben promover la temprana integración de sus hijos y dar a conocer, tanto a familiares como a otros miembros de la comunidad, la naturaleza genética de la enfermedad y su carácter no contagioso7.
Ictiosis severa en el neonatoLos neonatos que nacen con membrana colodión o eritrodermia son de alto riesgo neonatal debido a una insuficiencia cutánea severa7,13. Los avances en la terapia intensiva neonatal han sido claves para mejorar el pronóstico de las ictiosis congénitas severas14.
El uso de incubadoras humidificadas (50-70% de humedad) evita la inestabilidad térmica y la deshidratación hipernatrémica. Por otra parte, el apoyo nutricional precoz permite enfrentar el aumento de la demanda metabólica secundaria al alto recambio epidérmico, evitando la desnutrición13.
La piel debe ser estrechamente vigilada por el alto riesgo de infecciones. La higiene es esencial para evitar la sobreinfección y favorecer la eliminación de las escamas. Los hidratantes, como la vaselina, deben aplicarse regularmente, ya que sustituyen parcialmente la función de barrera, permiten la descamación y proporcionan un ambiente apropiado para la curación de fisuras y erosiones7,13. La indicación de retinoides sistémicos se reserva para neonatos con ictiosis arlequín, ya que se ha postulado que su uso precoz podría aumentar la sobrevida de estos pacientes14.
Futuras terapiasLa terapia génica ha surgido como una posible alterativa curativa en el tratamiento de las ictiosis, basándose en la corrección de los genes defectuosos. Estudios experimentales en ictiosis severas, como IA e IL, muestran resultados prometedores. En IA se ha logrado corregir las mutaciones en el gen ABCA12, recuperando la expresión fenotípica de queratinocitos en cultivos celulares15, mientras que en IL se ha conseguido restaurar la enzima TGM-1, corrigiendo la expresión fenotípica de piel humana trasplantada en ratones inmunosuprimidos16,17.
ConclusionesLas ictiosis son un grupo heterogéneo de enfermedades secundarias a un trastorno de la cornificación, que tienen un gran impacto en la calidad de vida de los pacientes y sus familias. La nueva clasificación de ictiosis ha permitido una mejor comprensión de la enfermedad y un correcto diagnóstico, ya que considera los aspectos clínicos, histológicos y moleculares de esta enfermedad. El descubrimiento de los trastornos genéticos subyacentes es de suma importancia para el desarrollo de terapias más específicas y eventualmente curativas, como la terapia génica.
Conflicto de interesesEste trabajo cumple con los requisitos sobre consentimiento/asentimiento informado, comité de ética, financiación, estudios animales y sobre la ausencia de conflicto de intereses según corresponda.
