



El síndrome de Turner (ST) es un trastorno genético asociado con anormalidades del cromosoma X, que ocurre un 1 caso por cada 2,000 a 2,500 mujeres nacidas vivas. Se asocia generalmente con talla baja, disgenesia gonadal y niveles circulantes insuficientes de esteroides sexuales que conducen a insuficiencia ovárica e infertilidad. El manejo de la terapia hormonal de reemplazo (THR) en la adolescente, es aún un tema controversial. La terapia con estrógenos está disponible en diversas formulaciones y diferentes regímenes, que requieren estudios de las diferencias en los efectos sobre la salud y seguridad.
Este artículo revisa la literatura médica sobre terapia hormonal de reemplazo en pacientes con ST. Se realizó una búsqueda en la base de datos de PubMed y en las principales revistas de endocrinología, concentrando los artículos publicados de ST con especial énfasis en la literatura reciente.
Por el tipo de estudios publicados, el número de pacientes involucrados, el tiempo de seguimiento y la diversidad de esquemas de tratamiento utilizados; fue difícil tener una comparación objetiva, por lo que consideramos imprescindible la realización de estudios prospectivos multicéntricos que permitan formular guías de atención de las pacientes con ST.
The Turner syndrome (TS) is a genetic disorder associated with abnormalities in the chromosome X, which occur in 1 out of 2,000 to 2,500 female live births. Usually is associated with low size, gonad digenesis and insufficient circulating levels of sex steroids leading to ovarian failure and infertility. The use of the hormonal replacement therapy (HRT) in the adolescents is still a controversial topic. Estrogen therapy is available in many formulations and different regimes, requiring studies of differences in the effects on health and safety.
This article reviews the literature on HRT in patients with TS. A search was conducted in the PubMed database and the main magazines of Endocrinology, concentrating TS published articles with special emphasis on recent literature.
By the type of published studies, the number of patients involved, time tracking and the diversity of schemes of treatment used; it is difficult to have an objective comparison, so multicenter prospective studies that allow to formulate guidelines is essential.
El síndrome de Turner es un trastorno cromosómico que se caracteriza por la presencia de un número variable de cambios en el fenotipo y en los órganos internos, siendo la disgenesia gonadal y la talla baja los hallazgos más típicos. El síndrome es causado por la ausencia parcial o completa de un cromosoma X en al menos un tejido del organismo1.
Historia y terminologíaEn 1938 Henry H. Turner describió en siete mujeres un cuadro caracterizado por estatura baja, ausencia de caracteres sexuales femeninos, pterigium colli y cubitus valgus; él consideró que una alteración hipofisaria era la causa responsable de la falta de función ovárica de estas pacientes. Previamente, Otto Ullrich había descrito el mismo cuadro clínico en 1930. En 1959 Ford et al comunicaron la pérdida de uno de los cromosomas X en las pacientes con síndrome de Turner.
PrevalenciaLa prevalencia descrita en el mundo es de 1 X 2,000 a 1 X 5,000 recién nacido vivos del sexo femenino. Cerca del 1 a 2% de todas las concepciones presentan una monosomía X; de ellas 99% terminan en abortos espontáneos, generalmente durante el primer trimestre del embarazo2. En México se desconoce la prevalencia exacta. En Noviembre del 2011 la Asociación del Síndrome de Turner de México A.C. contaba con 300 pacientes inscritos3.
DiagnósticoEl diagnóstico se establece por las diversas características clínicas, puede sospecharse prenatalmente por la presencia de hallazgos ultrasonográficos específicos como edema fetal, aumento en la translucencia nucal e higroma quístico; también pudiera encontrarse coartación de la aorta, anomalías renales, polihidramnios y retardo en el crecimiento intrauterino, además de los resultados del triple o cuádruple marcador materno (alfa feto proteína, hGC, inhibina A y estriol no conjugado)4. Después del nacimiento del 20 al 30% de las pacientes afectadas son diagnosticadas por sus características fenotípicas; en la adolescencia el 35% por la baja estatura; además se pueden encontrar niveles elevados de FSH y ausencia de características sexuales secundarias. En México el diagnóstico se suele realiza en promedio entre los 6 y 12 años de edad3.
Cuando se sospecha el diagnóstico de ST debe realizarse un cariotipo. Cuando realizamos el estudio cromosómico convencional en cultivo de sangre periférica, cerca del 50% de los casos muestran una monosomía X (45,X). Otros cariotipos que se encuentran en el ST son mosaicismos con otras líneas celulares, tales como 46,XX o 46,XY ó 47,XXY. Las anomalías estructurales del cromosoma X son también frecuentes tales como isocromosoma de brazos largos del cromosoma X, deleciones, anillos o translocaciones.
Pacientes con cariotipo 45X0 tienen más características clínicas que aquellos que tienen mosaicismo con una línea celular normal; pero en los casos de mosaicismo, es importante investigar la presencia de una línea celular que tenga el cromosoma Y, el cual se encuentra en aproximadamente en el 5% de los casos, ya que cuando este cromosoma está presente existe un riesgo incrementado de 7-10% de desarrollar un gonadoblastoma y disgerminoma en la glándula disgenésica, por lo que se recomienda una gonadectomía profiláctica5.
Algunos autores consideran que todos los fetos viables son mosaicos y que la inactivación del cromosoma X faltante ocurre después de la fertilización. En general el ST no es familiar, y no existe mayor riesgo para las mujeres que ya tuvieron un hijo (abortado o vivo) con este síndrome de tener otro hijo con el mismo padecimiento.
Modificaciones hormonalesLas modificaciones hormonales presentes en el ST derivan principalmente de la alteración de la función ovárica. El proceso de diferenciación del ovario que ocurre en el feto con ST inicia normalmente en la 4ª semana de gestación, las células germinales migran a las crestas genitales y a la semana 18 de gestación inicia una degeneración prematura de los folículos del ovario, los que son reemplazados por abundante tejido conectivo (estrías gonadales) lo que resulta en falla ovárica.
En el momento de la pubertad no se produce el aumento de los niveles estrogénicos, por lo que no se produce el crecimiento y maduración del útero, vagina y mamas. Las hormonas hipofisiarias que controlan la función ovárica, LH y particularmente FSH están aumentadas. Este aumento es ya aparente en los primeros meses de vida, como resultado de la involución ovárica. Durante la infancia, entre los 3 y los 7 años, los niveles de LH y FSH son normales, aunque después de la administración de Gn-RH se observa un aumento de ambas en mayor grado que en sujetos normales. Después de los 8 años de vida los niveles comienzan a aumentar nuevamente por encima de la normalidad; por ello, a esta edad, la determinación de las gonadotropinas puede tener valor diagnóstico. El mecanismo de feedback entre el ovario y los centros reguladores hipófisis e hipotálamo esté intacto, lo que se demuestra por el hecho de que las gonadotropinas pueden reducirse a niveles normales con el tratamiento sustitutivo con estradiol y progesterona.
La tasa de estradiol es muy variable y no se correlaciona con la tasa basal de gonadotropinas. Los niveles de andrógenos suprarrenales son normales y se elevan en la adrenarquia, pero por encima de los 15 años los niveles de testosterona, androstenediona y dehidroepiandosterona son bajos a causa de la hipofunción ovárica.
A mediados de la década de los 80's se sugirió que la disminución del crecimiento en las pacientes con ST se relacionaba con una baja producción de GH al final de la niñez y principio de la adolescencia; sin embargo, es probable que sea debido a la falta del aumento inducido de la secreción de GH por esteroides sexuales en el periodo puberal. También podría influir en este sentido el exceso de peso que suelen presentar estas pacientes. Parece ser poco probable que exista un déficit en la capacidad para sintetizar IGF-1 o de responder a la IGF-I ya que la secreción de este inducida por GH parece ser normal; así como la sensibilidad celular a la IGF-16.
Unos niveles de hormona del crecimiento adecuados que están asociados a una producción normal de esteroides sexuales, son necesarios para que ocurra el episodio de crecimiento que se presenta durante la pubertad y que es conocido como el estirón puberal.
Fenotipo - clínicaLa mayoría de los pediatras están familiarizados con las características clínicas clásicas del ST, por lo que el diagnóstico se sospecha sobre todo por la talla baja, linfedema de manos y pies, cuello alado, línea de implantación del cabello baja en el cuello y cúbito valgo. En la tabla 1 se muestran algunas de las anomalías clínicas y la frecuencia con que se presentan.
Fenotipo del síndrome de Turner
| Hallazgo | Frecuencia % |
|---|---|
| Estatura baja | 100 |
| Infertilidad | 97-99 |
| Fallo ovárico prematuro | 90-96 |
| Manos y pies hinchados | 80 |
| Tórax ancho | 80 |
| Mamas hipoplasicas y muy separadas | 80 |
| Línea posterior del cabello baja | 80 |
| Rotación y forma anormal de las orejas | 80 |
| Mandíbula inferior baja | 70 |
| Brazos hacia el exterior en su unión con el hombro | 70 |
| Uñas débiles y dobladas hacia arriba | 70 |
| Osteopenia, Osteoporosis y fracturas a edades tempranas | 50-80 |
| Dedos anulares cortos | 60 |
| Hipertensión arterial | 50 |
| Nevos pigmentados | 50 |
| Cubito valgo | 47 |
| Anormalidades cardiacas | 45-55 |
| Cuello corto | 40 |
| Anomalías renales | 39-70 |
| Bóveda del paladar estrecha | 38-50 |
| Obesidad | 30 |
| Sordera neurosensorial | 27-40 |
| Intolerancia a carbohidratos | 15-50 |
| Hipotiroidismo | 15-30 |
| Estrabismo | 18 |
| Escoliosis | 10 |
| Gonadoblastoma | 7-10 |
| Diabetes mellitus tipo 2 | 5-10 |
| Alopecia | 2-5 |
| Vitíligo | 3-5 |
| Psoriasis | < 5 |
| Artritis reumatoide juvenil | < 5 |
| Enfermedad celiaca | 4-6 |
La presentación clínica varía con la edad. Desde el periodo de la infancia es muy característico la talla baja, motivo por el que en toda niña con talla corta debe considerarse en el diagnóstico diferencial el ST, sobre todo si se acompaña de soplo cardiaco. El retraso de la menarquia con talla corta debe considerarse como ST mientras no se demuestre lo contrario. La presencia de vello axilar y púbico no debe considerarse como evidencia de pubertad, pues se deben a la presencia de andrógenos de origen adrenal7. En las mujeres adultas con talla corta, con infertilidad o irregularidades en la menstruación debe descartarse ST. No obstante algunas mujeres con ST tienen menarquia, Da Silva Negreiros y cols.8 evaluaron 123 pacientes y encontraron que la pubertad espontanea ocurrió en el 78.4% de las pacientes, y el 21.6% requirió terapia estrogénica para presentar la pubarca. Aunque en otras series se ha visto que únicamente el 15-30% presentan pubertad espontánea. La conclusión es que todas las pacientes con ST necesitan estrógenos exógenos.
La mayoría de las pacientes con ST tienen inteligencia normal (IQ 90), aunque pueden existir trastornos de aprendizaje, sobre todo en lo que se refiere a la percepción espacial, lectura, coordinación visual-motora y matemáticas. También pueden presentar hiperactividad, inmadurez, ansiedad y depresión9. Lepage10 encontró en un estudio de 46 pacientes con ST y un grupo control de 45 pacientes que las mujeres con ST, jóvenes y adolescentes presentaban una reducción del volumen de materia gris, materia blanca y área de superficie sobre la corteza parietal y occipital que pudiera ser consecuencia de la deficiencia de estrógenos. Las características clínicas varían según la anomalía citogenética que presenta la paciente con ST. Los hallazgos clínicos característicos los presentan las pacientes con monosomía X, y de la variedad con isocromosoma del Xq; los pacientes con deleción del Xp presentan sobre todo estatura corta y malformaciones congénitas y aquellas con deleción de Xq a menudo solo presentan disgenesia gonadal.
Calidad de vidaEn un estudio Mexicano que incluyo 45 pacientes con ST de edades entre 2 y 42 años se encontró un ausentismo escolar y laboral en 51.1% de las pacientes siendo la causa más común la necesidad de atención médica (46.7%); un 57.8% expresaron que han sido objeto de burla de compañeros de trabajo, escuela o familiares; 15.5% refirieron sentirse insatisfechas con su vida y, a pesar de esto, 60% no buscó apoyo en un grupo de ayuda. En la población total se refirió un gasto mensual alto para medicamentos relacionados con su atención, sin incluir el costo de la hormona de crecimiento, la cual fue recibida en algún momento de su vida en el 73.3% de las pacientes; mientras que 70.3% de las mayores a 14 años recibieron terapia con estrógenos3.
En un estudio en población con hipogonadismo congénito, donde se incluyeron 26 pacientes con Sx de Turner y 62 pacientes con hipogonadismo por otras causas, se encontró que las pacientes con ST tuvieron menor excitación que la población general, pero las pacientes con otra causa de hipogonadismo presentaron aún menor excitación sexual, orgasmo y dolor11.
TratamientoEl tratamiento de las pacientes con ST requiere la valoración y seguimiento periódico a diferentes edades. Además de ser un manejo multidisciplinario; como ya se ha mencionado anteriormente nosotros nos centraremos en la terapia hormonal de reemplazo.
Tratamiento de la talla bajaLa terapia con hormona del crecimiento (GH) cual fue aprobada por la FDA en 1996. La talla de las pacientes sajonas con ST en promedio es de 145cm. Con la GH se obtiene una talla final media de 1.50 y con GH más oxandrolona 152.1, casi 6cm más que los controles históricos12.
La aceleración de la velocidad de crecimiento es contemporánea al inicio puberal, seguida por una fase de enlentecimiento hasta el cierre de los cartílagos de crecimiento. Durante la pubertad la ganancia en talla es de aproximadamente 20cm. Las niñas con ST no presentan el estirón puberal, lo que sumado a su talla baja previa, explica los 20cm bajo la media de la talla familiar.
La terapia con hormona del crecimiento recombinante humana debe de iniciar tan pronto como la talla este por debajo de la percentil 5 para la edad. Se aplica por vía subcutánea en una dosis de 0.375mg/kg por semana.
La talla adulta depende de la talla al inicio del tratamiento, respuesta en el primer año, talla genética, edad al inicio del tratamiento, dosis media semanal del GH, edad al finalizar el tratamiento, uso de oxandrolona, cuya aplicación aun está en debate y peso al nacimiento. El cariotipo no ha mostrado ninguna influencia13.
Los efectos adversos de esta terapia incluyen hipertensión intracraneal, escoliosis, deslizamiento de la epífisis de la cabeza femoral y metabolismo anormal de glucosa.
Tratamiento del hipogonadismoEl objetivo de la terapia hormonal de reemplazo es simular la pubertad normal, incluyendo las siguientes metas:
- •
Inducir y mantener un desarrollo sexual y sangrado uterino cíclico.
- •
Permitir un pico de densidad mineral ósea adecuada para prevenir la pérdida de masa ósea y la necesidad de tratamiento de osteopenia, osteoporosis y fracturas a futuro.
- •
Mantener un adecuado desarrollo de la piel y musculatura14.
- •
Mantener y desarrollar la memoria relacionada a la organización y planteamiento de metas futuras (Memoria de trabajo)15.
- •
Prevención de enfermedades cardiovasculares
- •
Factor protector de riesgos para cáncer Endometrial y ovárico.
La inducción de la pubertad comienza a la edad de 11-12 años para tratar de simular lo mayor posible a la pubertad natural. La edad de inicio puede ser anterior o posterior en función de factores individuales. Para la edad de inicio se busca el equilibrio entre momento óptimo para el ámbito psicológico, la formación de hueso y la estatura. Antes de iniciar la inducción de la pubertad se tendrán que determinar LH y FSH para excluir maduración tardía.
La formulación optima de estrógenos, dosis, vía de administración y tiempo de inicio de progesterona es controversial. Al inicio se comienza con una dosis baja de estrógeno, que durante un período de 2-4 años se elevara a la dosificación para adultos. La mayoría de los expertos en el tema recomiendan que se inicie con 1/10 a 1/8 de la dosis de sustitución del adulto, seguido de un aumento gradual en un periodo de 2-4 años.
El efecto de los estrógenos es en realidad bifásico, por una parte estimulan el crecimiento en bajas dosis (1/10-1/8 de la dosis de sustitución adulta), y por otra parte, a altas dosis (sobre 50pmol/lt [1.3 ng/dL]) son inhibidores del crecimiento16.
Para la sustitución de estrógenos hay diferentes preparaciones disponibles. Típicamente se utiliza la forma oral del 17 beta estradiol; esta preparación es idéntica a la de estradiol de origen natural y tiene los efectos benéficos de los estrógenos; es seguro de usar y no tiene efectos colaterales sobre los parámetros metabólicos de los factores de coagulación y los niveles de lipoproteínas de alta densidad (HDL), así como aumentar la densidad ósea. La ventaja de la preparación transdérmica es que no tiene ningún efecto de primer paso en el hígado, causando menos inducción de proteínas hepáticas. Puede utilizarse estrógenos equinos conjugados, pero su formulación contiene múltiples sustancias estrogénicas con diferentes potencias biológicas que pueden no ser indicadas en esta población.
Hay controversia de cual vía es la más adecuada: en un estudio de Torres-Santiago y Taboada17 en donde participaron 40 pacientes con ST de 16.7 ± 1.7 años de edad, se encontró que ambos grupos tuvieron concentraciones de estradiol normales, después de 6 a 12 meses el porcentaje corporal de grasa, la DMO y oxidación de lípidos fueron similares en ambos grupos. El IGF-1 fue más bajo con el estradiol oral, pero la supresión de gonadotropinas fue comparable. El estriol y SHBG fueron más altas en el grupo de terapia oral. La vía transdérmica resultó más fisiológica que la vía oral. En otro estudio el estradiol transdérmico resultó en mayor DMO, así como mayores incrementos en la talla final y el volumen uterino18.
La administración oral de estradiol conduce a la concentración farmacológica de la hormona en la vena portal antes de que se metabolice en el hígado; este primer paso resulta en un aumento de la producción hepática de globulinas transportadoras de varias hormonas, factores de coagulación, lipoproteínas y angiotensinógeno, efectos que son evitados vía transdérmica19.
A continuación presentamos algunos de los esquemas más utilizados, aunque, como hemos mencionado, no hay un consenso y cada experto utiliza algún esquema según su propia experiencia:
1. Administración oral de 17 beta estradiol: tabletas 0.5mg 17 β-estradiol.
Esquema:
- •
1er año: 5 mcg/kg/día
- •
2° año: 7.5 mcg/kg/día
- •
3er año: 10 mcg/kg/día
- •
4° año: 15 mcg/ kg/día
- •
Dosis de adultos: 1-2mg/día
Esquema de dosificación transdérmica20:
- •
Los primeros 0-12 meses se aplica el parche antes de dormir para eliminarse a la mañana siguiente.
- –
< 40kg: 1/8 del parche de 25 mcg (= 3.1 mcg).
- –
40-55kg: 1/6 del parche de 25 mcg (= 4.2 mcg).
- –
> 55kg: ¼ de parche 25 mcg (= 6.2 mcg).
- –
- •
12-18 meses: aplicar antes de dormir, retirar la mañana siguiente:
- –
Si se inicia el primer año con 1/8 aumentar a 1/6 del parche de 25 mcg (= 4.2 mcg).
- –
Si ha iniciado con 1/6, aumentar a ¼ del parche de 25 mcg (= 6.2 mcg).
- –
Si se inicia con ¼ pase a la mitad del parche de 25 mcg (= 12.5 mcg).
- –
- •
18-30 meses:
- –
Si se usó a los 12-18 meses 1/6 parte del parche de 25 mcg, pasar a ¼ (= 6.2 mcg).
- –
Si se administró ¼ usar la mitad del parche (= 12.5 mcg).
- –
Si durante los 12-18 meses se administro la mitad, utilizar ¾ del parche (= 18.8 mcg).
- –
- •
30-36 meses: antes de dormir cortar el parche de 25 mcg en dos partes iguales y colocar ambas partes, por la mañana quitar una parte y solo antes de dormir volverla a colocar.
- •
36 meses
- –
2 veces por semana parche de 50 mcg, o
- –
1 parche por semana de 75-100 mcg
- –
Método de administración: el parche debe colocarse en una parte no dañada de la piel, en un lugar que no sea demasiado móvil (parte superior de glúteos, abdomen, espalda), colocar el nuevo parche en otra parte de la piel.
2. Terapia con Etinilestradiol: presentación en cápsulas.
- •
Año 1: 5 mcg/kg/día
- •
Año 2: 7.5 mcg/kg/día
- •
Año 3: 10 mcg/kg/día
- •
Año 4: 15 mcg/kg/día
- •
Dosis para adultos: 1-2mg por día
Vía transdérmica con Gel de 17 beta estradiol:
- •
mg el primer an¿o.
- •
mg el segundo an¿o.
- •
0.5mg el tercer an¿o.
- •
mg el cuarto an¿o.
- •
1.5mg el quinto an¿o.
Se puede iniciar con dosis de estradiol en parche de 5-7 mcg/kg, se puede tomar determinación sérica de estradiol después de 1-2 semanas lo que hace posible ajustar la dosis individualmente; si el desarrollo mamario inicia dentro de los primeros 3 meses se debe considerar reducir la dosis21.
En un estudio donde participaron 50 niñas de entre 12 y 14 años en tratamiento con GH y datos de insuficiencia ovárica (FSH elevada) se les indujo el desarrollo puberal utilizando en la mitad de ellas una dosis individualizada de 5-15 mcg/kg por día durante 2 años y en el otro grupo se utilizó una dosis fija de 0.2mg diarios durante el primer año seguido de 0.5mg diarios durante el segundo año, se observó que el inicio del desarrollo de caracteres sexuales medido por escala de Tanner inicio antes en el grupo de dosis fijas (733 días) vs el grupo de dosis individualizado (818 días) (p = 0.046), la edad ósea no mostró aceleración inadecuada y no hubo alteración en la estatura esperada en ninguno de los grupos22.
Tratamiento con progestágenosEste tratamiento se comienza dos años después de iniciar la terapia con estrógenos, o antes si se produce spotting. Debe coincidir idealmente con una ecografía ginecológica que muestre un útero > 55mm y un endometrio mayor de 3mm.
Nuevamente se prefieren los progestágenos naturales; se dan durante 10-14 días al mes, sobre todo si se desea hemorragia por deprivación. Los esquemas más utilizados son:
- •
Didrogesterona (Duphaston®) 5-10mg/día del día 1 al 14 del mes, con estrógenos continuos. Es el más utilizado porque es lo más similar a la progesterona natural. Puede dar la hinchazón como efecto secundario.
- •
Progesterona micronizada (Geslutin®) (idéntica a la progesterona), la dosis es generalmente 200mg por día por la noche antes de acostarse, si fuera necesario, 100mg por la mañana, del día 1 al 10, con estrógenos continuos.
- •
Medroxiprogesterona (Provera®) de 10mg por 10 días al mes con estrógenos continuos.
- •
Noretisterona 5mg. (Primolut®-Nor) Los estudios en animales han demostrado que tiene ligero efecto residual androgénico.
Al prescribir la terapia de reemplazo hormonal se puede hacer la distinción entre una administración bifásica con estrógeno continuo y progestina durante 14 días por ciclo, y el esquema continuo, con el uso de estrógenos y progestágenos de manera continua. El uso continuo produce un riesgo de sangrado irregular, especialmente durante el primer año de uso de un 12-22% frente a 8% que producen los bifásicos23.
Efectos de reemplazo estrogénico a baja dosis durante la infanciaComo ya mencionábamos anteriormente la disgenesia ovárica que afecta al 90% de nuestras pacientes con ST resulta en una deficiencia estrogénica desde la infancia y que resulta en efectos perjudiciales en diversos tejidos por lo que en los últimos años se han llevado a cabo estudios de los efectos del reemplazo estrogénico utilizando ultra bajas dosis.
En Indianápolis se realizó un estudio en 123 pacientes: 62 de ellas recibieron placebo y las 61 restantes recibieron 0.025 mcg/kg/día (25 ng) de los 5 a los 8 años; y 0.05 mcg/kg/día de los 8 a los 12 años; estas dosis fueron reducidas al 50% en menores de 12 años que presentaron desarrollo mamario, sangrado vaginal y en menores de 14 años que tuvieron un avance rápido en la edad ósea: las niñas que recibieron estrógenos tuvieron una telarca más temprana (11.6 vs 12.6 años) y un ritmo más lento de pubertad (3.3 vs 2.2 años), ambos grupos tuvieron menarca tardía (15 años), además de los efectos mencionados en la capacidad cognitiva y la ganancia de estatura mediada por hormona de crecimiento24.
En un Estudio de Kodama y cols.25, que incluyó 67 pacientes, se utilizó una dosis de adulto en 27 pacientes mostrando una densidad mineral ósea (DMO) mayor que los 30 pacientes tratados con bajas dosis de estrógenos, y los 10 pacientes sin terapia estrogénica (0.808g/cm2, 0.174g/cm2 y 0.664g/cm2 respectivamente). El incremento anual de DMO fue significativamente mayor cuando se iniciaron estrógenos a dosis de adultos antes de los 18 años, 4.4% antes de los 18 años vs 3.1% después de los 18 años.
Anticonceptivos oralesLos anticonceptivos orales tienen como desventaja que no hay reemplazo de estrógeno en la semana de descanso. En esta semana suele producirse una hemorragia por deprivación que normalmente se inicia en el segundo o tercer día después de la última tableta que contiene el ingrediente activo.
Con una paciente 45 XO sin el desarrollo puberal y la menarquia espontánea la posibilidad de embarazo es casi nula; en consecuencia, después de la inducción de pubertad no se recomienda ninguna indicación de anticonceptivos orales y debe continuar con terapia de reemplazo hormonal.
En las mujeres con mosaico que tuvieron pubertad espontánea y tienen su propio ciclo, aunque es raro, se pueden utilizar anticonceptivos orales cuando no hay deseo de tener hijos. Si en la semana de descanso surgen síntomas menopáusicos, tales como sofocos y sudores nocturnos, se llevará a cabo una evaluación de la función de la reserva ovárica y se considerara un cambio a la preparación continua.
Aquellos adolescentes que presentan un aumento del riesgo de trombosis y/o una coagulopatía no se recomienda el uso de anticonceptivos, pero si es factible el uso de THR por vía transdérmica asociado a progestágenos en forma cíclica26.
Las píldoras de combinación con 30 microgramos de etinilestradiol proporcionan mayor control del ciclo que las píldoras combinadas que contienen 20 microgramos, las cuales dan un mayor riesgo de sangrado irregular y metrorragia. La nueva píldora con valerato de estradiol/ dienogest (Qlaira®) en comparación con una píldora de 20 mcg de etinilestradiol/levonorgestrel muestra relevantes ventajas en términos de control del ciclo27.
Riesgos de la terapia hormonal de reemplazo en pacientes con síndrome de TurnerNo se cuenta con estudios significativos sobre los riesgos y efectos secundarios de los anticonceptivos hormonales y la terapia de reemplazo hormonal en mujeres con síndrome de Turner. Casi todas las recomendaciones se basan en estudios hechos en pacientes sanas. Es necesario tomar en cuenta que pacientes con ST están expuestas a la terapia estrogénica durante décadas. Básicamente serían los mismos riesgos de uso a cualquier edad, dependiendo del estrógeno utilizado, la dosis y la vía de administración.
Hasta el momento no se han reportado pacientes con cáncer de mama en los estudios realizados. Existe el riesgo de hiperplasia endometrial por uso de Estrógenos, pero este riesgo es controlable con progestágenos cíclicos.
El riesgo de tromboembolismo venoso con los anticonceptivos orales en mujeres jóvenes es pequeño, el riesgo absoluto por la píldora es de 3.6 por cada 10,000 mujeres por año, el cual varía con la preparación, principalmente por el tipo de progestágeno. Una preparación que contiene levonorgestrel o noretindrona (píldora de segunda generación) da el menor riesgo relativo de trombosis (OR 3.6 y 3.9 respectivamente).
Los anticonceptivos orales con diferentes progestágenos da un mayor riesgo de trombosis: gestodeno (OR: 5.6), linestrenol (OR: 5.6), norgestimato (OR: 5.9), drospirenona (OR: 6.3), acetato de ciproterona (OR: 6.8) y desogestrel (OR: 7.3)28.
Cuanto más se acerca el progestágeno a la fórmula estructural de la progesterona pura (21 carbonos = didrogesterona, medroxiprogesterona), se ve un menor efecto en la glucosa y el metabolismo de los lípidos. Derivados de la testosterona (19 carbonos = linestrenol, noretindrona) tienen un efecto anabólico androgénico mayor en comparación con la progesterona pura29.
Existe la posibilidad de que el uso de etinilestradiol en las dosis habitualmente requeridas pueda provocar hipertensión arterial, ya que su degradación llevaría a la producción de radicales libres que potenciarían el aumento de los sustratos de renina circulantes.
Pacientes que presenten intolerancia a la lactosa son susceptibles de ser tratadas con vías alternativas a la oral, ya que un comprimido de estrógenos y/o progesterona contiene alrededor de 50-75mg de lactosa14.
En pacientes con ST hay un aumento de los niveles séricos de enzimas hepáticas, y los resultados de diversos estudios son contradictorios: encontrando en algunos de estos estudios que los niveles están disminuidos por la THR. En un estudio en el cual se utilizo 17-beta estradiol vía oral, la capacidad hepática de conversión de galactosa, indocianina verde y antipirina fueron normales y no se modificaron por la THR30.
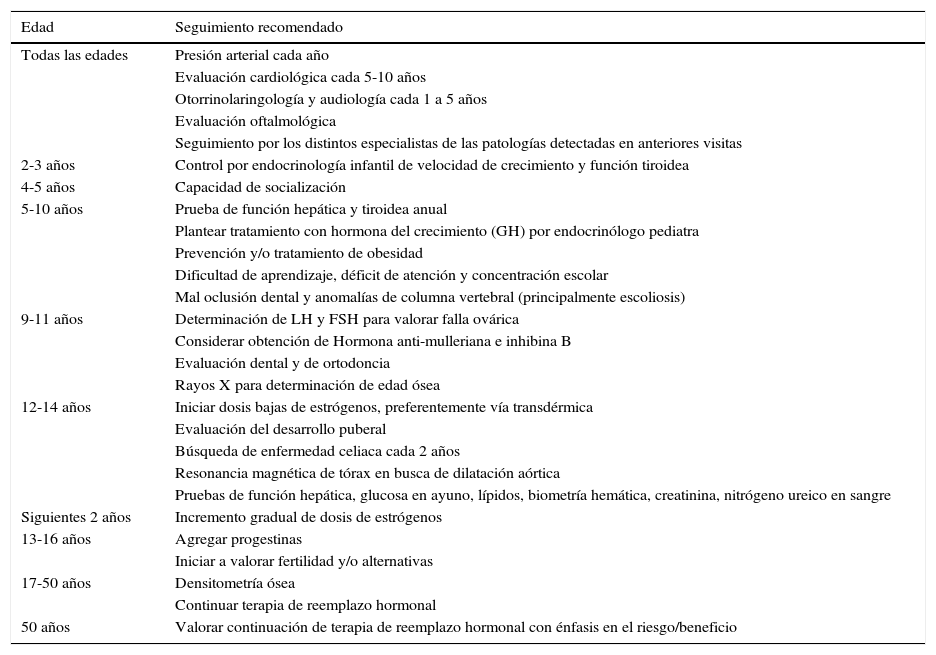
El seguimiento indicado de las pacientes con síndrome de Turner en THR se muestra en la tabla 2.
Seguimiento de las pacientes con síndrome de Turner
| Edad | Seguimiento recomendado |
|---|---|
| Todas las edades | Presión arterial cada año |
| Evaluación cardiológica cada 5-10 años | |
| Otorrinolaringología y audiología cada 1 a 5 años | |
| Evaluación oftalmológica | |
| Seguimiento por los distintos especialistas de las patologías detectadas en anteriores visitas | |
| 2-3 años | Control por endocrinología infantil de velocidad de crecimiento y función tiroidea |
| 4-5 años | Capacidad de socialización |
| 5-10 años | Prueba de función hepática y tiroidea anual |
| Plantear tratamiento con hormona del crecimiento (GH) por endocrinólogo pediatra | |
| Prevención y/o tratamiento de obesidad | |
| Dificultad de aprendizaje, déficit de atención y concentración escolar | |
| Mal oclusión dental y anomalías de columna vertebral (principalmente escoliosis) | |
| 9-11 años | Determinación de LH y FSH para valorar falla ovárica |
| Considerar obtención de Hormona anti-mulleriana e inhibina B | |
| Evaluación dental y de ortodoncia | |
| Rayos X para determinación de edad ósea | |
| 12-14 años | Iniciar dosis bajas de estrógenos, preferentemente vía transdérmica |
| Evaluación del desarrollo puberal | |
| Búsqueda de enfermedad celiaca cada 2 años | |
| Resonancia magnética de tórax en busca de dilatación aórtica | |
| Pruebas de función hepática, glucosa en ayuno, lípidos, biometría hemática, creatinina, nitrógeno ureico en sangre | |
| Siguientes 2 años | Incremento gradual de dosis de estrógenos |
| 13-16 años | Agregar progestinas |
| Iniciar a valorar fertilidad y/o alternativas | |
| 17-50 años | Densitometría ósea |
| Continuar terapia de reemplazo hormonal | |
| 50 años | Valorar continuación de terapia de reemplazo hormonal con énfasis en el riesgo/beneficio |
Es importante inducir y mantener el desarrollo puberal, cuya ausencia genera un serio impacto psicológico en la adolescente con ST. Se ve enfrentada a la falta de cambios físicos característicos, a la ausencia de menarquia y menstruaciones que en muchas sociedades, desde un punto de vista cultural, sería el pilar su femineidad.
Se debe hacer prevención de enfermedades cardiovasculares, reducción de la mortalidad por causa cardiovascular como la mejora del patrón del perfil lipídico.
El abordaje de las pacientes debe ser integral e incluir historia clínica completa, examen físico exhaustivo, estudios de imagen, medición de hormonas y cariotipo con bandeo G de alta resolución. En ciertas pacientes con síndrome de Turner existen situaciones especiales en las que es necesario realizar hibridación fluorescente in situ para detectar fragmentos del cromosoma Y; si este es el caso, debe complementarse con resonancia magnética nuclear y resección de estría gonadal debido al alto riesgo de gonadoblastoma5.
Es necesario el impulso de investigaciones más amplias y estandarizadas para poder llegar a resultados más confiables y que se puedan comparar entre sí con el fin de realizar guías para el mejor diagnóstico, manejo y seguimiento de las pacientes con Síndrome de Turner.
También es importante una mayor información sobre este síndrome a la población general a fin de identificar más tempranamente a las pacientes afectadas, ofrecerles un tratamiento oportuno, crear grupos de apoyo para mejorar la calidad de vida y concientizar sobre la importancia de continuar con la terapia hormonal de reemplazo hasta inicios de la 6ta década de la vida, ya que muchas pacientes al dejar de llevar un seguimiento con el endocrinólogo pediatra abandonan el tratamiento por falta de información y orientación adecuada.