

Desde 1965, la Directiva 65/65/CEE1 modificada por la Directiva 87/21/CEE2, en su artículo 4.8 apartado a) punto iii) ya se refiere a la posibilidad de comercializar medicamentos esencialmente similares a un medicamento innovador. A pesar de no utilizar la terminología de «medicamento genérico», define el mismo concepto para esencialmente similar, dos medicamentos que tienen la misma composición cuantitativa y cualitativa en principio activo, la misma forma farmacéutica y son bioequivalentes. El medicamento innovador (original o de referencia) debe cumplir dos requisitos para poder servir de modelo al medicamento esencialmente similar:
El procedimiento de mutuo reconocimiento se utiliza para obtener una autorización sanitaria de comercialización en distintos países
de la Unión Europea
La primera autorización en la comunidad europea debe haber sido obtenida desde hace más de 6/10 años (aunque en España el período de protección de datos está establecido en 6 años, para 2001 se prevé que todos los países de la Unión Europea armonicen su legislación nacional para permitir la protección de datos durante 10 años). A este primer medicamento innovador autorizado se le denomina medicamento original.
Debe estar comercializado en el estado miembro donde se solicita la autorización de comercialización para el medicamento esencialmente similar. Esta versión del medicamento original autorizado en este estado miembro se le denomina medicamento de referencia.
En España, la referencia legislativa a la especialidad farmacéutica genérica no se encuentra hasta 1996 en la Ley 13/1996, de 30 de diciembre, de Medidas Fiscales, Administrativas y de Orden Social3. Dicha ley introduce a través de su artículo 169 modificaciones en distintos artículos de la Ley 25/1990, de 20 de diciembre, del Medicamento; en concreto, modificó los artículos 8, 16, 90 y 94 de la Ley del Medicamento.
El artículo 8 punto 6 bis comprende la definición de especialidad farmacéutica genérica como «la especialidad con la misma forma farmacéutica e igual composición cualitativa y cuantitativa en sustancias medicinales que otra especialidad de referencia, cuyo perfil de eficacia y seguridad esté suficientemente establecido por su continuo uso clínico. La especialidad farmacéutica genérica debe demostrar la equivalencia terapéutica con la especialidad de referencia mediante los correspondientes estudios de bioequivalencia. Las diferentes formas farmacéuticas orales de liberación inmediata podrán considerarse la misma forma farmacéutica siempre que hayan demostrado su bioequivalencia».
En los artículos 16, 90 y 94 se describe la denominación de una especialidad farmacéutica genérica, se introduce la sustitución de especialidades farmacéuticas genéricas por parte del profesional sanitario y se hace referencia la financiación por parte del Sistema Nacional de la Salud, respectivamente.
Posteriormente a la Ley 13/1996, la Dirección General de Farmacia y Productos Sanitarios (DGFyPS) publicó para la industria farmacéutica dos circulares, la Circular 3/97 de procedimientos de tramitación de solicitudes de especialidades farmacéuticas genéricas4, y la Circular 24/97 de inclusión de las siglas EFG y para obtener la condición de especialidades farmacéuticas genéricas5, ambas con la finalidad de concretar algunos aspectos descritos en ley.
En concreto, la Circular 3/97 describe las condiciones que deben cumplir las especialidades farmacéuticas genéricas para ser autorizadas, las partes de que consta el expediente de registro y los procedimientos de registro. La Circular 24/97 describe cómo disponer las siglas EFG y cómo obtener la condición de especialidad farmacéutica genérica.
Condiciones de autorización de las EFG
Los medicamentos genéricos deben cumplir exactamente las mismas exigencias que el medicamento innovador a las que se refiere el artículo 10 de la Ley 25/1990 del Medicamento: calidad, seguridad y eficacia, además de estar correctamente identificadas y acompañadas de información precisa y transparente. Pero además se les exige el cumplimiento de otros requisitos más específicos de su condición.
El medicamento genérico debe tener la misma composición cualitativa y cuantitativa en principio activo y con la misma forma farmacéutica que el medicamento innovador que toma de referencia, considerando a todas las formas farmacéuticas orales sólidas de liberación inmediata como una misma forma farmacéutica (cápsulas y comprimidos).
El principio activo no debe estar desaconsejado para formar parte de una especialidad farmacéutica genérica ya sea por tener un estrecho margen terapéutico o unas características especiales de biodisponibilidad. Estos principios activos son los que determine el Ministerio de Sanidad y Consumo a través del artículo 90.3 de la Ley 25/1990 del Medicamento y del artículo 11.6 del Real Decreto 767/936.
A pesar de que no hay mayores exigencias de formulación del medicamento, se deben tener en cuenta otros factores que pueden afectar a la equivalencia del medicamento genérico con el medicamento innovador. Por ejemplo, otras características fisicoquímicas del principio activo, el uso de excipientes diferentes o una forma distinta de fabricación son factores que suelen ser diferentes pero que no deben conducir a un medicamento que difiera significativamente del medicamento innovador en seguridad y eficacia.
La seguridad y eficacia del medicamento genérico deben estar avaladas por un uso clínico continuado del principio activo. En concreto, deben haber transcurrido 10 años desde que se autorizó el medicamento innovador en España o bien el medicamento debe estar autorizado como especialidad farmacéutica genérica en algún país de la Unión Europea en el que hubiera sido posible obtener la protección de una patente de producto para el principio activo. La calidad del medicamento genérico se debe demostrar con la presentación del Drug Master File (DMF) y los correspondientes procesos de validación.
Otra condición es que el medicamento innovador tomado de referencia ha de estar autorizado en España en base a un expediente de registro completo, es decir, que debe contener estudios de calidad, estudios farmacotoxicológicos y estudios clínicos llevados a cabo durante el proceso de investigación y desarrollo del medicamento. El medicamento genérico también realiza estudios de calidad, pero no aporta estudios farmacotoxicológicos ni clínicos, sino que debe realizar los estudios de bioequivalencia. Mediante los estudios de bioequivalencia se puede evaluar si la acción terapéutica entre el medicamento genérico y el medicamento innovador es lo bastante parecida como para utilizar alternativamente uno u otro considerándolos esencialmente similares.
Finalmente, la especialidad farmacéutica genérica se debe identificar con la Denominación Oficial Española (DOE), o de no existir, con la Denominación común Internacional (DCI) del principio activo, seguida del nombre o marca del laboratorio o fabricante y las siglas EFG.
Expediente de registro de una EFG
El expediente de registro consiste en un dossier elaborado por la compañía farmacéutica donde figura información exhaustiva acerca del medicamento genérico, con el objetivo de demostrar ante las autoridades sanitarias condiciones suficientes de seguridad, eficacia, calidad y pureza, identificación e información precisa y transparente.
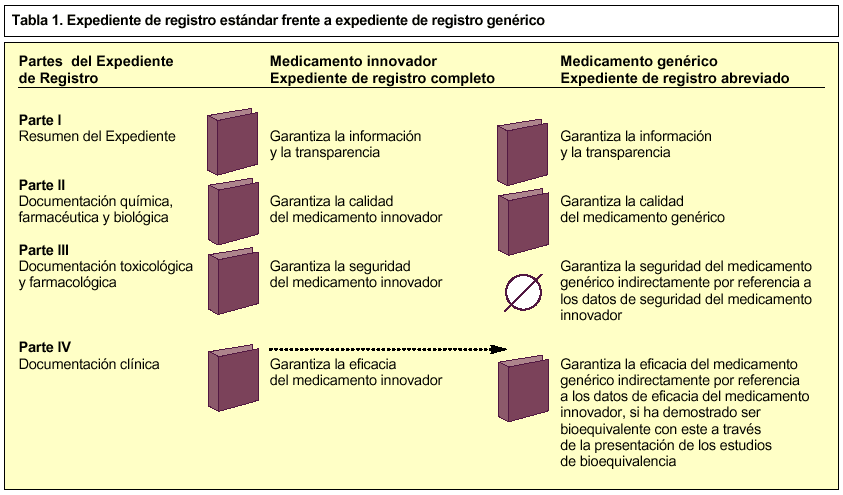
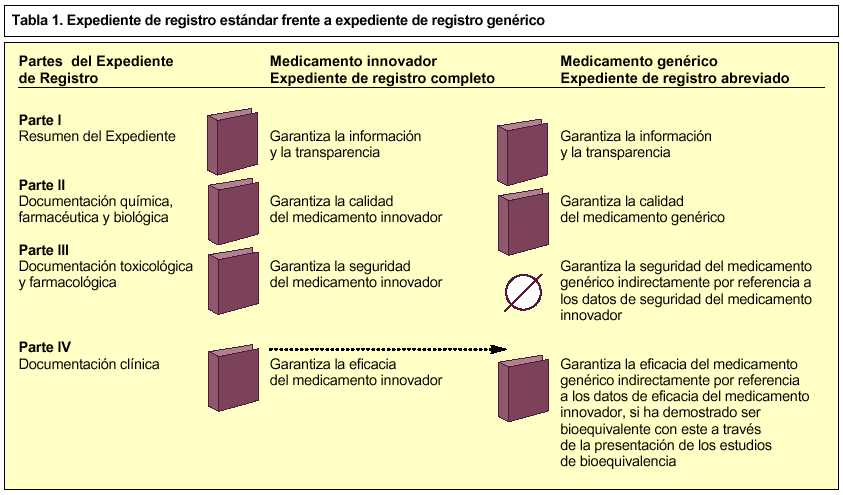
Los expedientes de registro de EFG se consideran expedientes abreviados, porque no contienen todas las partes de un expediente de registro estándar7, sino que hacen mención a información contenida en el dossier de un medicamento innovador en lo que se refiere a la parte III (documentación farmacotoxicológica) y parte IV (documentación clínica), para demostrar su seguridad y eficacia respectivamente (tabla 1). Esto significa que un dossier de un medicamento genérico está siempre vinculado con el dossier del medicamento innovador.
La financiación pública de las especialidades farmacéuticas bioequivalentes, como son los medicamentos genéricos, se ve afectada por el sistema de los precios de referencia
Así, el expediente de registro de una especialidad farmacéutica genérica consta de cuatro partes, aunque la tercera, en este caso, no procede.
Primera parte
Consiste en un resumen de todo el dossier y garantiza la información y la transparencia.
Segunda parte
En ella se debe aportar la documentación química, farmacéutica y biológica que garantice la calidad del medicamento.
Tercera parte
No procede.
Cuarta parte
Se debe aportar documentación sobre los estudios de bioequivalencia realizados según las normas vigentes sobre medicamentos de la Unión Europea.
Procedimientos de registro de una EFG
La tramitación de un expediente de registro hasta la autorización final por parte de las autoridades sanitarias de una especialidad farmacéutica genérica se puede llevar a cabo por dos procedimientos distintos: un procedimiento nacional y un procedimiento de ámbito europeo en el que intervienen las autoridades sanitarias de distintos países miembros, denominado procedimiento de mutuo reconocimiento.
Procedimiento de registro nacional
El procedimiento de registro nacional se utiliza para obtener una autorización sanitaria de comercialización en un determinado país de la Comunidad Europea, como puede ser España. En este tipo de procedimiento, la compañía farmacéutica demuestra la equivalencia de su medicamento genérico con el medicamento de referencia propio del país. La autorización concedida por la autoridad sanitaria del país sólo es válida en ese único país.
En España el procedimiento está contemplado en el Real Decreto 767/1993, de 21 de mayo6, modificado por el Real Decreto 2000/1995, de 7 de diciembre8. La Agencia Española del Medicamento (AEM) y la DGFyPS son los dos organismos activos en el procedimiento de registro nacional. La AEM actúa en primera instancia con la evaluación, autorización, registro y control de medicamentos mientras que se deja la competencia de la fijación de precios y la gestión del Sistema Nacional de Salud a la DGFyPS.
La parte II del expediente de registro, formada por documentación química, farmacéutica y biológica del medicamento genérico, es estudiada en la AEM por la División de Química y Tecnología Farmacéutica. En el caso de medicamentos genéricos biotecnológicos, la División de Productos Biológicos y Biotecnología también se integra en el proceso de evaluación. De la evaluación de la parte II y su informe de experto integrado en la parte I del expediente de registro, se emite de un informe de calidad.
La parte IV del expediente de registro formada por los estudios de bioequivalencia, es estudiada en la AEM por la División de Farmacología y Evaluación Clínica. De la evaluación de la parte IV y su informe de experto integrado en la parte I del expediente de registro, se emite un informe de evaluación. Esta división también es responsable de la evaluación de la información que la compañía propone dirigir a los profesionales sanitarios o ficha técnica, y de la información dirigida a los pacientes y consumidores, o prospecto y etiquetado, integrados en la parte I del expediente de registro.
Ambos informes, el de calidad y el de evaluación son estudiados por el Comité de Evaluación de Medicamentos de Uso Humano (CODEM). La autorización sanitaria de comercialización para un medicamento genérico es concedida o denegada por la AEM en base al informe emitido por el Comité de Evaluación de Medicamentos de Uso Humano y es válida únicamente en el territorio nacional.
Procedimiento de mutuo reconocimiento
El procedimiento de mutuo reconocimiento se utiliza para obtener una autorización sanitaria de comercialización en distintos países de la Unión Europea. En este tipo de procedimiento, la compañía farmacéutica demuestra la equivalencia del medicamento genérico con el medicamento de referencia de un sólo país llamado estado miembro de referencia. Una vez obtenida la autorización de comercialización en aquel estado miembro, las demás autorizaciones en los estados miembros concernidos se obtienen por reconocimiento de los diferentes estados miembros de la primera autorización. Este procedimiento está establecido en la Directiva 75/319 del consejo, de 20 de mayo, con sus modificaciones posteriores9.
Todavía hoy existen muchos problemas en la aplicación práctica de este procedimiento.
Uno de estos problemas se encuentra en el contenido de las fichas técnicas. Normalmente no coincide el contenido de la ficha técnica aprobada para el medicamento genérico en el estado miembro de referencia con el contenido de la ficha técnica en los medicamentos de referencia de los distintos estados miembros concernidos. Este hecho genera muchos problemas en el momento de reconocer la primera autorización y ello hace que se opte por el procedimiento nacional.
Otro problema se encuentra en el período de protección de datos. Existen diferencias en los distintos estados miembros y esto supone un problema cuando se pretende iniciar un procedimiento de mutuo reconocimiento tomando como estado miembro de referencia un país donde el período de protección de datos es inferior que en los estados miembros concernidos en los cuales no será posible el procedimiento hasta que no expire el período de protección.
Un medicamento genérico puede ser autorizado para todas las indicaciones terapéuticas, todas las formas farmacéuticas, las dosis y las posologías ya autorizadas al medicamento de referencia, aunque algunas de ellas estén autorizadas hace menos de 6/10 años, según Sentencia del Tribunal de Justicia de las Comunidades Europeas (Sala Quinta), de 3 de diciembre de 1998, en la que se señala que el plazo de protección 6/10 años no es aplicable a cada una de las indicaciones y posologías autorizadas para el medicamento original, sino al medicamento en sí mismo.
Una vez autorizado el medicamento como especialidad farmacéutica genérica, se debe asegurar el mantenimiento de la calidad del producto para garantizar una buena seguridad y eficacia.
Disposición de las siglas EFG
El artículo 16 de la Ley 25/1990 del Medicamento establece que «cuando se trate de especialidad farmacéutica genérica, la denominación estará constituida por la denominación oficial española o, en su defecto, por la denominación común o científica acompañada del nombre o marca del titular o fabricante. Las especialidades farmacéuticas genéricas se identificaran por llevar la sigla EFG en el envase y etiquetado general».
La sigla EFG debe disponerse en el embalaje exterior, en el acondicionamiento primario, en el cupón precinto, en el prospecto y en la ficha técnica en la misma línea y a continuación de la denominación del medicamento. Se escribe siempre en mayúsculas, con un tamaño superior al 50% del tamaño de la denominación y no deben ir al lado del nombre o marca del titular o fabricante ni del código nacional, ni de las siglas ASSS o cualquier otro símbolo.
La sigla EFG permite a los profesionales sanitarios diferenciar los medicamentos genéricos de todo el resto de especialidades farmacéuticas existentes en el mercado español. Por tanto, aunque existen especialidades farmacéutica denominadas con el nombre del principio activo seguido del nombre del fabricante, no son consideradas especialidades farmacéuticas genéricas si no llevan las siglas EFG. Este es el caso de los llamados «falsos genéricos», productos que se han comercializado imitando a medicamentos originales desamparados por una patente. La diferencia con los medicamentos genéricos es que no han tenido que demostrar bioequivalencia con el medicamento innovador.
Obtención de la condición de EFG
En el supuesto de las especialidades farmacéuticas autorizadas o en trámite de registro que no tengan la condición de especialidad farmacéutica genérica y quieran obtenerla, pueden conseguirlo mediante uno de los siguientes procedimientos:
Para especialidades farmacéuticas autorizadas con denominación genérica, el laboratorio puede solicitar la condición de especialidad farmacéutica genérica si demuestra cumplir con todos los requisitos establecidos en la Circular 3/97 «condiciones que deben cumplir las especialidades farmacéutica genéricas para ser autorizadas», excepto el requisito de llevar las siglas EFG.
Para especialidades farmacéuticas autorizadas con denominación fantasía, el laboratorio puede solicitar la calificación de EFG, si además de demostrar que cumple con todos los requisitos establecidos en la Circular 3/97, solicita un cambio de denominación de fantasía a nombre genérico.
Para especialidades farmacéuticas autorizadas con denominación genérica o de fantasía que no cumplen con todos los requisitos establecidos en la Circular 3/97, el laboratorio puede solicitar la calificación de EFG, si aporta documentación adicional que le permita cumplir con todos los requisitos establecidos en la circular y solicita, si procede, un cambio de denominación de fantasía a nombre genérico.
Para especialidades farmacéuticas en trámite de registro con denominación genérica que cumplen con todos los requisitos establecidos en la Circular 3/97, serán directamente autorizadas con la condición de especialidad farmacéutica genérica.
Para especialidades farmacéuticas en trámite de registro con denominación de fantasía, que cumplen con todos los requisitos establecidos en la Circular 3/97, el laboratorio puede solicitar un cambio de denominación de fantasía a nombre genérico y ser autorizadas con la condición de especialidad farmacéutica genérica.
Fijación del precio y financiación por parte del SNS
El precio es uno de los aspectos más relevantes de los medicamentos genéricos. Debido al hecho de que demuestran su seguridad y eficacia en base a un medicamento innovador, los costes de investigación preclínica y clínica son prácticamente nulos quedando entonces reducidos a la inversión en los estudios de desarrollo farmacéutico. Esta reducción de gastos hace que los medicamentos genéricos pueden salir al mercado a un precio inferior al de los medicamentos innovadores.
La fijación de precios y la gestión del Sistema Nacional de Salud (SNS) son competencia de la DGFyPS y tienen lugar una vez concedida, por parte de la AEM, la autorización sanitaria de comercialización. Entonces la compañía farmacéutica inicia los trámites para la fijación de precios según lo establecido en el Real Decreto 271/1990, de 23 de febrero10, y los trámites para su financiación por el SNS, según lo previsto en el Real Decreto 83/1993, de 22 de enero11.
Además, la financiación pública de las especialidades farmacéuticas bioequivalentes, como son los medicamentos genéricos, se ve afectada por el sistema de los precios de referencia. La Ley 13/1996, de 30 de diciembre3 y la Ley 66/1997, de 30 de diciembre12 introdujeron modificaciones en el artículo 94 de la Ley 25/1990 del Medicamento para establecer las bases de este sistema de precios. El Real Decreto 1.035/1999, de 18 de junio13, desarrolló ambas leyes para su aplicación práctica.
La Ley 13/1996 abrió la posibilidad al Gobierno para limitar la financiación pública de medicamentos estableciendo que, de entre las distintas alternativas bioequivalentes disponibles, sólo fueran financiadas las especialidades farmacéuticas cuyos precios no superaran un precio fijado para cada principio activo. Esta limitación en la financiación no excluía la posibilidad que el paciente eligiera otra especialidad farmacéutica prescrita por el médico de precio superior, siempre pagando la diferencia.
Un año después, la Ley 66/1997 estableció la obligación de sustituir la especialidad farmacéutica prescrita que supere el precio de referencia por un medicamento genérico de igual composición cualitativa y cuantitativa, forma farmacéutica, vía de administración y dosificación y de igual o inferior precio que el establecido.
El Real Decreto 1035/1999, de 18 de junio, desarrolló esta normativa y creó el sistema actual de precios de referencia. En este real decreto se describe como se agrupan e identifican las especialidades, como se calculan los precios de referencia y como se deben sustituir las especialidades:
El Ministerio de Sanidad y Consumo determina los conjuntos homogéneos, grupos de especialidades farmacéuticas calificadas como bioequivalentes y que por lo menos una de ellas sea especialidad farmacéutica genérica. Así, todas las especialidades integradas en un conjunto homogéneo son intercambiables entre sí porque tiene la misma composición cualitativa y cuantitativa, misma forma farmacéutica, dosis, vía de administración, y son equivalentes terapéuticamente. Las especialidades farmacéuticas integradas en los conjuntos homogéneos se identifican por llevar las siglas EQ en el cupón precinto.
El precio de referencia es la cuantía máxima que se financiará de las especialidades farmacéutica que estén incluidas en un conjunto homogéneo. Los precios de referencia de nuevos conjuntos homogéneos son aprobados con una frecuencia mínima anual.
Por último, la sustitución de una especialidad farmacéutica bioequivalente de precio superior al de referencia, debe hacerse por una especialidad farmacéutica genérica del mismo conjunto homogéneo con un precio igual o inferior al de referencia, excepto si el médico acompaña la prescripción con un informe en el que justifique la no sustitución por motivos de alergia, intolerancia o otra incompatibilidad del paciente al cambio de excipientes.
Más tarde, el Real Decreto-Ley 5/2000, de 24 de junio14, en su artículo 2.2 incidió notablemente en el precio de las especialidades farmacéuticas y de forma particular en las especialidades farmacéuticas genéricas. Para estas últimas pasa de un margen profesional para las oficinas de farmacia por su dispensación y venta al público de un 27,9% a un 33% del precio de venta al público (PVP) sin impuestos para las presentaciones con un precio de venta del laboratorio (PVL) igual o inferior a 78,34 euros (13.035 pesetas). Para el resto de las presentaciones, es decir, las de PVL superior a 78,34 euros (13.035 pesetas), establece un margen de 33,54 euros (5.580 pesetas) por envase según la norma general.
Presente y futuro
A pesar del intento del gobierno de destacar la misma calidad, seguridad y eficacia que otro medicamento de marca y el beneficio en referencia al precio, el consumo de genéricos avanza muy lentamente. Las principales causas de ello son:
La implantación de los precios de referencia ha provocado que los medicamentos con marca hayan rebajado sus precios para poder competir con los medicamentos genéricos.
En España, la Ley de Patentes no permite empezar los estudios de desarrollo de genéricos hasta la caducidad de la patente, mientras que hay países donde si se permite. Esto hace que los desarrollos de genéricos se trasladen a otros países.
La falta de información a pacientes, médicos y farmacéuticos.
Todo ello conlleva a que en la actualidad España sea uno de los países con menor implantación de medicamentos genéricos, situándose su consumo alrededor del 5% del total de medicamentos a diferencia de la media Europea que se sitúa alrededor del 15%. En relación a otros países europeos esta cifra resulta aún más baja, ya que por ejemplo, Alemania y Reino Unido alcanzan cifras cercanas al 50%.
Bibliografía y notas
1.Directiva 65/65/CEE del Consejo, de 26 de enero, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas sobre especialidades farmacéuticas (DOCE de 9 de febrero).
2.Directiva 87/21/CEE del Consejo, de 22 de diciembre de 1986, por la que se modifica la Directiva 65/65/CEE relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas sobre especialidades farmacéuticas (DOCE de 17 de enero de 1987).
3.Ley 13/1996, de 30 de diciembre, de Medidas Fiscales, Administrativas y del Orden Social. Artículo 169 (BOE del 31).
4.Circular de la DGFyPS n.º 3/97 de procedimiento de tramitación de solicitudes de especialidades farmacéuticas genéricas.
5.Circular de la DGFyPS n.º 24/97 de inclusión de las siglas EFG y para obtener la condición de especialidad farmacéutica genérica.
6.Real Decreto 767/93, de 21 de mayo, por el que se regula la evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso humano fabricados industrialmente (BOE de 2 de julio).
7.Montpart E, Martín MP. Registro de Especialidades y Productos sanitarios (II). El Procedimiento de Registro Nacional. Offarm 2001;20(3):134-42.
8.Real Decreto 2.000/95, de 7 de diciembre, por el que se modifica el Real Decreto 767/93, de 21 de mayo, que regula la evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso humano fabricados industrialmente (BOE de 12 de enero de 1996).
9.Directiva 75/319/CEE del Consejo, de 20 de mayo, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas sobre medicamentos (DOCE de 9 de junio).
10.Real Decreto 271/1990, de 23 de febrero, sobre reorganización de la intervención de precios de los medicamentos de uso humano (BOE de 2 de marzo).
11.Real Decreto 83/1993, de 22 de enero, por el que se regula la selección de los medicamentos a efectos de su financiación por el Sistema Nacional de Salud (BOE de 19 de febrero).
12.Ley 66/1997, de 30 de diciembre, de Medidas fiscales, Administrativas y del Orden social (BOE del 31).
13.Real Decreto 1.035/1999, de 18 de junio, por el que se regula el sistema de precios de referencia en la financiación de medicamentos con cargo a fondos de la Seguridad Social o a fondos estatales afectos a la Sanidad (BOE del 29).
14. Real Decreto-Ley 5/2000, de 23 de junio, de Medidas Urgentes de Contención del Gasto Farmacéutico Público y de Racionalización del Uso de los Medicamentos (BOE del 24).