




La encefalopatía enpongiforme bovina, más conocida por la enfermedad de las vacas locas, no sólo está causando un notable impacto social, sino también una gran expectación entre la comunidad científica por la aparición de un nuevo agente infeccioso: los priones. El gran número de artículos publicados en la prensa no especializada ha originado un cierto clima de alarma social, que afecta directamente a diversos sectores relacionados con el ganado vacuno. La aparición de la enfermedad
en los humanos debido a la ingesta de tejido nervioso de animales infectados ha convertido un descubrimiento científico
en un gran problema de salud pública.
La encefalopatía enpongiforme bovina (BSE) pertenece a un grupo de enfermedades llamadas encefalopatías espongiformes transmisibles (EET). Se caracterizan por la aparición de cavidades en las neuronas, dando un aspecto esponjoso al encéfalo y se hace muy evidente en cortes histológicos de los cerebros afectados.
Son neuropatías de infección lenta, es decir, tienen un período de incubación muy largo de hasta 20-30 años, aunque una vez aparecen los primeros síntomas (aprehensión progresiva, hiperestesia y descoordinación del paso), la enfermedad progresa rápidamente y en el período de un año se produce inevitablemente la muerte.
La mayoría de los científicos han aceptado la existencia de unas partículas proteicas, llamadas priones, como los agentes responsables directos de las encefalopatías espongiformes.
Estas proteínas se caracterizan por haber sufrido modificaciones, adquiriendo la capacidad de autogeneración, pero carecen de ácidos nucleicos (ADN o ARN), considerados hasta ahora elementos básicos necesarios para la replicación de cualquier agente vivo.
La evidencia de que una proteína tenga capacidad de autogeneración y pueda transmitir una enfermedad infecciosa ha causado gran sorpresa entre la comunidad científica.
Historia
En 1957 fue descrita una rara enfermedad, llamada Kuru, que afectaba principalmente a las mujeres y niños de algunas tribus de Nueva Guinea. Es una enfermedad degenerativa del sistema nervioso central, que comienza a manifestarse con un andar inseguro, pérdida de coordinación, temblores, demencia y finalmente termina con la muerte. El Kuru se transmitía a través de un ritual caníbal que formaba parte de la ceremonia fúnebre de estas tribus. Cuando alguien moría, el encéfalo era comido por niños y mujeres, y a lo largo de los años padecían la enfermedad. En cambio, los hombres no participaban de esta práctica y por este motivo raramente eran afectados por esta enfermedad. Cuando esta práctica fue interrumpida, el Kuru desapareció casi por completo.
Posteriormente se demostró que el Kuru era una enfermedad muy parecida a otra enfermedad neurodegenerativa mortal que afectaba a los seres humanos de todo el mundo, descrita en Alemania en 1920 y conocida con el nombre de Creutzfeldt-Jakob (CJ). La enfermedad de CJ produce depresión, alteraciones del comportamiento, alucinaciones y demencia.
Años más tarde se describieron otras enfermedades humanas parecidas a la enfermedad de CJ y fueron designadas como encefalopatías espongiformes, como la enfermedad de Gertmann Sträussler Scheinken o la llamada enfermedad de insomnio familiar letal.
El grupo de encefalopatías espongiformes humanas son poco frecuentes, siendo la más común la enfermedad de CJ, que presenta una incidencia de 1/1.000.000/año.
En los animales, la encefalopatía más común era el scrapie o prurito lumbar de los ovinos y caprinos. Los animales afectados por esta encefalopatía empiezan por tener dificultades al andar y perder la capacidad de coordinación de los movimientos, temblores, convulsiones, irritabilidad y picores que lleva a los animales a rascarse hasta tal punto de arrancarse el pelo y la piel.
La BSE es de origen reciente y parece que fue provocada por la modificación en el tratamiento de los piensos utilizados para alimentar el ganado vacuno.
En 1985 se observó por primera vez en una granja del sur de Inglaterra, una vaca adulta con síndrome neurológico que fue descrito como «hipersensibilidad crónica con síndrome de descoordinación»; 7 meses después en el mismo rebaño se dieron nueve casos más con los mismos síntomas clínicos. En noviembre 1986, en el Laboratorio Veterinario Central del Ministerio de Agricultura, Pesca y Alimentos del Reino Unido fue identificada la enfermedad conocida con el nombre de la BSE. El análisis histopatológico de estos animales mostraba una gran similitud con los cerebros de las ovejas infectadas por scrapie.
La causa de la nueva enfermedad fue atribuida a la alteración, pocos años antes, del método de fabricación de las harinas alimenticias preparadas a partir de los despojos de origen ovino rechazados en los mataderos. A mediados de los años ochenta, la BSE empezó a manifestarse en el ganado vacuno que había sido alimentado con estas harinas, y entre 1986 y 1991 murieron más de 28.000 bovinos en el Reino Unido.
El Gobierno británico, a modo de prevención, prohibió en 1988 el uso de suplementos alimenticios derivados de despojos de otros rumiantes para la fabricación de piensos. Aun así, en 1997 se contabilizaron 170.000 casos de BSE en el Reino Unido.
La importancia de la BSE, a parte de las nefastas consecuencias económicas para la industria de la explotación del ganado vacuno, aumentó al considerarse el riesgo potencial que constituía para el hombre. En 1995 aparecieron los dos primeros casos relacionados con la BSE. Desde entonces, 27 personas han muerto de lo que se conoce actualmente como la variante nueva de la enfermedad de CJ, cuyo agente transmisible parece ser el mismo agente causante de la BSE (Pattison, 1998).
Encefalopatías espongiformes
La enfermedad de las vacas locas, como ya se ha explicado anteriormente, pertenece al grupo de las encefalopatías espongiformes trans misibles (TSE). Éstas son enfermedades neurodegenerativas que pueden afectar tanto a los animales como al hombre. La etiología de las TSE, es decir, su transmisión en la cadena biológica, es desconocida en la mayoría de los casos. Su transmisión puede ser horizontal o vertical, y en algunos casos existen factores genéticos implicados.
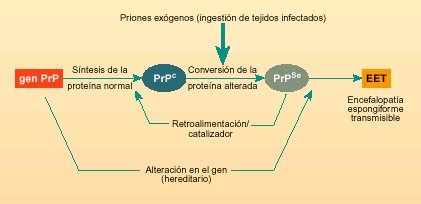
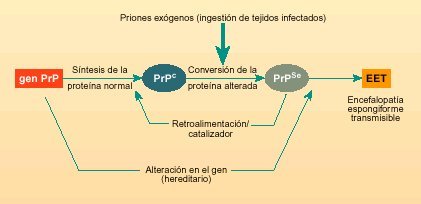
Fig. 1. Vías posibles de conversión de la glicoproteína «normal» (PrPc) en la glicoproteína «alterada» (PrPSc o prión).
La patología de la BSE es muy parecida a la del scrapie de la oveja. Los cambios más notorios son la astrogliosis, la vacuolización intracelular, la pérdida de neuronas y la formación de placas amiloides ocasionales. La ausencia de variaciones observada en los patrones de vacuolización de los tejidos encefálicos en el ganado afectado, tanto procedente de casos naturales como de infecciones experimentales, sugiere la posibilidad de que una única cepa de prión pudiera ser la causante de la epidemia de BSE (Wells et al, 1992).
En la especie humana, algunas de las encefalopatías espongiformes son conocidas desde hace más de 100 años. Su aparición es muy rara y quizá sea esta la razón por la que han sido poco estudiadas.
La nueva variedad de la enfermedad de CJ, producida por el mismo agente infeccioso que la BSE, es de difícil diagnóstico debido a las claras diferencias que existen con la enfermedad de CJ clásicamente conocida. Se declara con un promedio de 26,7 años y tiene una evolución lenta (7-24 meses). Los primeros síntomas que aparecen son predominantemente psiquiátricos: ansiedad, depresión y cambios de conducta. Semanas o años después de la aparición de los primeros síntomas se manifiestan claras alteraciones en el cerebro, observándose trastornos en la coordinación muscular. Finalmente, se produce demencia y, consecuentemente, la muerte.
Actualmente se conoce muy poco sobre las encefalopatías espongiformes, sobre todo las humanas. Ni tan siquiera se conoce bien cuál es y cómo actúa el agente infeccioso responsable de las EET. La mayor parte de los científicos aceptan la hipótesis de los priones, es decir, la existencia de unas proteínas que han sido modificadas y que han adquirido la capacidad de autorreproducirse. Aun así, existen otras hipótesis como la que defiende un grupo de investigadores de la Universidad de Yale (Estados Unidos), que han observado que en algunas de estas EET existe la presencia de un retrovirus: un virus con ARN.
Los priones
La hipótesis de los priones fue propuesta por Prusiner en 1982, época en la que aún no se conocía el origen de esta proteína. En un artículo publicado en 1991, Prusiner señalaba: «Introduje el término prión para que las partículas proteicas infecciosas que son la causa del scrapie, de la enfermedad de CJ, de la GSS y del Kuru se distingan de los viroides y de los virus.» Actualmente, los priones se designan como proteínas infecciosas con capacidad de autogeneración, al igual que las bacterias y los virus, pero con la extraña diferencia de que no poseen ni ADN ni ARN, considerados hasta ahora indispensables para la replicación de un agente infeccioso.
Uno de los problemas a que se enfrentan los científicos es el hecho que los priones no se destruyen mediante agentes físicos como temperaturas elevadas, radiaciones ionizantes, o por tratamientos químicos, como las DNAsas (que degradan el DNA) o RNAsas (que degradan el RNA), y tampoco se ha encontrado ningún tipo de respuesta inmunitaria tal como sucede en la infección por virus y bacterias. Sólo se han obtenido resultados positivos con las proteasas, agentes que degradan las proteínas, indicando así la naturaleza del agente infeccioso.
En la última década se ha asociado la glicoproteína PrPc con los priones. La proteína PrPc se encuentra normalmente en la superficie de las células, especialmente en las neuronas, en las células de la glía y en los linfocitos, pero no se conoce su función fisiológica. La historia evolutiva de esta proteína es muy antigua y se encuentra en muchas especies de mamíferos. Parece ser que es necesaria para el funcionamiento de la sinapsis nerviosa y para la potenciación a largo plazo de la zona del hipocampo, en el encéfalo.
El prión o glicoproteína PrPSc, es la consecuencia de una alteración estructural producida en la glicoproteína PrPc. Este cambio en su estructura puede ser producido por dos mecanismos: una mutación en el gen que la codifica, o por el consumo de alimentos que contengan la proteína PrPSc (prión), ya que ésta podría causar la transformación de las proteínas celulares normales (PrPc) en proteínas alteradas (PrPSc).
Se sabe que la ingestión de encéfalos contaminados por priones puede ser una
de las causas de la transmisión de las EET
La forma alterada de esta proteína (PrPSc o prion), además de dañar las células nerviosas de manera directa, según se sugiere actualmente, también puede actuar como catalizador en la conversión de PrPc a PrPSc. El proceso de infección es semejante al de los virus y bacterias, pero no se debe a que el agente infeccioso se reproduzca, sino a la conversión de estas glicoproteínas normales a glicoproteínas alteradas o priones. Ello explicaría por qué las encefalopatías espongiformes transmisibles pueden presentarse esporádicamente y sin causas aparentes, ser hereditarias o resultar de contagio. Los tres casos implican la aparición inicial de una pequeña cantidad de PrPSc. En las formas esporádicas, la enfermedad se debería a la conversión espontánea de algunas moléculas en priones; en las hereditarias, sería debido a mutaciones en el gen, que se encuentra en el cromosoma 20 y que codifica para la proteína PrPc, facilitando la conversión a PrPSc; y en la adquiridas, el aporte inicial de PrPSc sería consecuencia del contagio (fig. 1).

En los tejidos cerebrales de humanos o de animales sanos no se han detectado la presencia de PrPSc o priones. Esto permite utilizar la proteína-prión como marcador de diagnóstico patológico.
Existen varias técnicas para su detección, como la inmunohistoquímica o la Western Blot, una técnica muy sensible y específica que consiste en la digestión de una parte del tejido cerebral con proteinasa K (agente que disgrega el tejido). El proceso es el siguiente: se realiza una extracción de las proteínas en general y la proteína-prión se separa de las otras proteínas mediante una electroforesis en gel de poliacrilamida; finalmente, se transfiere en una membrana de nitrocelulosa y se realiza un inmunoensayo que nos permitirá distinguir claramente los priones de las proteínas restantes.
Barreras intraespecíficas
Alrededor de un 10% de los casos de las encefalopatías espongiformes son hereditarios y la causa de la enfermedad se atribuye a una mutación en el gen que codifica para la glicoproteína PrPc o para una proteína alterada infecciosa (PrPSc o prión). El 90% de los casos restantes no son hereditarios. Este elevado porcentaje sugiere la existencia de más de una vía de transmisión de esta enfermedad. Se sabe que la ingestión de encéfalos contaminados por priones puede ser una de las causas de la transmisión de las EET, pero no se sabe cómo actúa en el sistema nervioso central del individuo afectado, ni cómo se transporta, ni cuál es su recorrido. Actualmente, sólo existen hipótesis y sugerencias.
El problema que más nos preocupa es saber si el prión que causa la EET en una especie que consigue ultrapasar la barrera interespecífica y afectar a otras especies, concretamente la humana. Actualmente se sabe que los priones que afectan a los mamíferos son diferentes en cada especie, es decir, el prión que afecta a la oveja es diferente al de la vaca y al del hombre. La secuencia de aminoácidos de la PrPc humana se diferencia de la ovina y la bovina en más de treinta posiciones, mientras que las de ovinos y bovinos difieren entre ellas en sólo siete posiciones. Esto podría explicar por qué el scrapie no se contagia de la oveja al hombre, pero en ciertas condiciones sí que podría transmitir a la vaca y generar la conocida BSE. En un principio, la diferencia en la composición química de la proteína humana a la de la vaca se consideró fundamental para la supuesta imposibilidad de transferir la BSE al hombre. Sin embargo, parece ser que existen ciertas regiones del prión vacuno y humano que son parecidas y que se constituyen en zonas cruciales para la infección. Así pues, esta hipótesis podría explicar la transmisión de la enfermedad de la BSE al hombre (Krakauer et al, 1996).
Conclusión
A pesar de todos los esfuerzos realizados en la investigación de las encefalopatías espongiformes, y más concretamente de la BSE, existen todavía numerosas lagunas en el conocimiento de éstas y su transmisión entre animales y humanos.
Actualmente, el sistema nervioso central es la única fuente de priones conocida. Los músculos, lo que habitualmente llamamos carne, así como la leche y sus derivados, no transmiten la enfermedad. Los considerados materiales de riesgo son, pues, los preparados alimenticios como el paté, las salchichas o las hamburguesas que pueden contener restos de tejido nervioso bovino.
Desde el pasado 1 de enero se han puesto en marcha las nuevas directivas europeas al respecto. Se han empezado a eliminar las harinas de carne para piensos de consumo animal y también se han empezado a realizar los análisis a los animales que se sacrifiquen para consumo con más de 2 años y medio de edad. La aparición del llamado «test rápido» asegurará que no se consuma ningún animal enfermo. Y en aquellos animales sospechosos de padecer la enfermedad de la BSE, se realizaran los ensayos específicos y confirmativos en el laboratorio y, en caso afirmativo, se procederá a su correcto procesamiento. *
Bibliografía general
Krakauer et al. Phylogenesis of prion protein. Nature 1996; 380: 675.
Pattison IH. A sideway look to the scrapie saga: 1732-1991. Prion diseases of human and animals. Prusiner, Collinge, Powell y Anderton (eds.). Londres: Ellis Horwood Publishers, 1992; 15-22.
Pattison J. The emergence of spongiform encephalopathy and related diseases. Emerging Infectious Diseases 1998; 4.
Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136-144.
Prusiner SB. The prion diseases. Scientific American 1995; 272: 48-57.
Scott MRD, Telling GC, Prusiner SB. Transgenetics and gene targeting in studies of prion disease. Current Topics in Microbiology and Immunology 1996; 207: 95-118.
Wells GAH, Hawkins SAC, Hadlow WJ, Spencer YI. The discovery of BSE and observations on the vacuolar changes. En: Prion diseases of human and animals. Prusiner, Collinge, Powell y Anderton (eds.). Londres: Ellis Horwood Publishers, 1992; 256-274.
Wilesmith JW, Wells GAH. Bovine spongiform encephalopathy. Current Topics in Microbiology and Immunology 1991; 172: 21-36.