





La farmacovigilancia es una responsabilidad compartida por las autoridades sanitarias, las compañías farmacéuticas y los profesionales sanitarios. Ante la nueva reglamentación aprobada mediante el RD 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, las autoras estudian la evolución y el funcionamiento actual del Sistema Español de Farmacovigilancia (SEFV).

Alos medicamentos se les exige calidad, seguridad y eficacia para que puedan ser comercializados y utilizados por la población. Los estudios realizados durante la investigación y el desarrollo de un nuevo medicamento proporcionan un buen conocimiento de su calidad y su eficacia; sin embargo, hay limitaciones para conocer su seguridad. Sólo su utilización en la población general y las condiciones de la práctica habitual permiten conocer con mayor precisión su perfil de seguridad. A este último aspecto se refiere la farmacovigilancia, que tiene por objeto proporcionar información sobre la seguridad de los medicamentos que ya se encuentran en el mercado, con el fin de asegurar que la relación beneficio/riesgo es favorable.
El Real Decreto 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, desarrolla la Ley del Medicamento e incorpora al ordenamiento jurídico español la normativa europea1. El importante alcance de su contenido nos conduce a la redacción de varios artículos interrelacionados. En este primero, se estudia la evolución y el funcionamiento actual del Sistema Español de Farmacovigilancia; en los siguientes, se analizarán con profundidad el papel de la industria farmacéutica y la intervención de las autoridades sanitarias cuando se presente un problema de seguridad; y por último, se tratarán los estudios postautorización.
Según el real decreto objeto de estudio, cabe entender por Sistema Español de Farmacovigilancia de medicamentos de uso humano la «estructura descentralizada que integra las actividades que las administraciones sanitarias realizan para recoger y elaborar la información sobre reacciones adversas a los medicamentos, coordinado por el Ministerio de Sanidad y Consumo, a través de la Agencia Española del Medicamento».
La Agencia Española del Medicamento (AEM), los órganos competentes en materia de farmacovigilancia de las comunidades autónomas y los profesionales sanitarios, constituyen en la actualidad el SEFV. El órgano integrado por la AEM y los órganos competentes en materia de farmacovigilancia de las comunidades autónomas forman el Comité Técnico del SEFV, encargado de unificar los criterios de funcionamiento y evaluar la información proporcionada por el SEFV.
Evolución legislativa
Aunque el objetivo de este artículo es el estudio del Real Decreto 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, consideramos interesante recordar a grandes trazos la evolución legislativa de lo que actualmente es el SEFV, ya que nos facilitará apreciar y comprender el alcance de la nueva reglamentación.
La primera norma que reguló la farmacovigilancia en España fue la Orden de 12 de noviembre de 1973, en la que se sentaban las bases para un sistema de farmacovigilancia organizado. Establecía la obligación de notificar de reacciones adversas al Centro Nacional de Farmacobiología para laboratorios farmacéuticos, médicos, farmacéuticos y servicios médicos, quirúrgicos y farmacéuticos hospitalarios, pero los resultados que se obtuvieron fueron muy pobres.
Tuvo que pasar casi una década para que en 1982 el Servicio de Farmacología Clínica de la Universidad Autónoma de Barcelona, con una ayuda económica del Fondo de Investigaciones Sanitarias de la Seguridad Social (FISSS), pusiera en marcha una experiencia piloto basada en la notificación voluntaria por tarjeta amarilla. A propuesta del Ministerio de Sanidad y Consumo, en 1983, la División de Farmacología Clínica se designó como centro de referencia español en el Programa Internacional de Farmacovigilancia de la OMS.
En 1985, se constituyó la Comisión Nacional de Farmacovigilancia como órgano consultivo del Ministerio de Sanidad y Consumo en materia de efectos adversos o tóxicos de los medicamentos
En 1984, la Dirección General de Farmacia y Productos Sanitarios (DGFyPS), con el asesoramiento de la OMS, realizó diversas actuaciones para establecer las bases de lo que hoy día constituye el SEFV. Para poner en marcha el sistema se eligió un camino descentralizado que, entonces comenzó a extenderse hacia el resto de las comunidades autónomas en las que se fueron creando centros regionales de farmacovigilancia.
En 1985, se constituyó la Comisión Nacional de Farmacovigilancia como órgano consultivo del Ministerio de Sanidad y Consumo en materia de efectos adversos o tóxicos de los medicamentos. Entre las funciones de la comisión figura la de aconsejar al Ministerio sobre la adopción de medidas para evitar o reducir los riesgos que comporta el uso de los medicamentos por la población. En 1997, esta comisión pasa a denominarse Comité de Seguridad de Medicamentos, nombre que conserva en la actualidad.
Un año después, en 1986, se aprueba la Ley General de Sanidad. De su articulado destacamos el artículo 95.5 en el que se indica que todas las personas calificadas que presten sus servicios en los servicios sanitarios y de investigación y de desarrollo tecnológico público tienen el derecho de participar y el deber de colaborar en la evaluación y el control de medicamentos y productos sanitarios; y el artículo 99, donde se expresa la obligatoriedad que tienen los importadores, fabricantes y profesionales sanitarios de comunicar a la autoridad sanitaria todas las reacciones adversas graves de que tengan noticia.
El programa en materia de farmacovigilancia se sometió a la consideración del recién creado Consejo Interterritorial del Sistema Nacional de Salud, órgano encargado de la coordinación de la Sanidad Nacional y formado por representantes de las 17 consejerías autonómicas.
Posteriormente, se aprobó la Ley del Medicamento que dedica el Capítulo Sexto del Título II a la Farmacovigilancia; lo integran dos artículos: el 57, relativo a la obligación de declarar, y el 58, destinado al SEFV. Concretamente, en su artículo 57.1 y con carácter de normativa básica, destaca el deber del profesional sanitario de comunicar con celeridad los efectos inesperados o tóxicos causados por medicamentos. Esta obligación se hace extensiva a los fabricantes y titulares de autorizaciones sanitarias de medicamentos. En el artículo 58.3, de forma expresa, se manifiesta la obligación de médicos, veterinarios, farmacéuticos, enfermeros y otros profesionales sanitarios de colaborar con el Sistema Español de Farmacovigilancia. De esta forma, se contempla la farmacovigilancia como una actividad multidisciplinaria.
El incumplimiento de la comunicación comporta una serie de infracciones y sanciones que se encuentran recogidas en el capítulo II, artículo 107 de la Ley del Medicamento. Estas infracciones se consideran graves (artículo 108), con una graduación en mínimo, medio y máximo, y son objeto de la sanción administrativa correspondiente (artículo 109) sin perjuicio de las responsabilidades civiles, penales o de otro orden que puedan concurrir.
El RD 767/1993, de 21 de mayo, por el que se regula la evaluación, la autorización, el registro y las condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso humano fabricados industrialmente, desarrolla la Ley del Medicamento e incorpora en nuestro ordenamiento jurídico la normativa europea. Este reglamento se vio notablemente modificado dos años más tarde mediante el RD 2.000/1995, de 7 de diciembre, debido a la aprobación del Reglamento del Consejo 2309/93/CEE por el que se crea la Agencia Europea para la Evaluación de Medicamentos y en el que se establece un sistema para mejorar la cooperación y el intercambio de información entre los estados miembros en relación con el control de los medicamentos y, en particular, con el control de sus reacciones adversas en condiciones normales de empleo por medio de los sistemas nacionales de farmacovigilancia.
La norma que estudiamos adecua la legislación española en materia de farmacovigilancia a lo establecido en la Directiva 2000/38 de la comisión, de 5 de junio, que introdujo notables cambios en la normativa europea; en concreto, el capítulo V bis «Farmacovigilancia» de la Directiva 75/319/CEE del Consejo, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas sobre especialidades farmacéuticas. En este punto cabe hacer una observación al contenido de lo expuesto en los antecedentes normativos que preceden al articulado del reglamento, las referencias a directivas comunitarias antes citadas, expresamente derogadas en la Directiva 2001/83/CE del Parlamento Europeo y del Consejo de 6 de noviembre, por la que se establece un Código Comunitario sobre medicamentos para uso humano y cuyo contenido se encuentra comprendido en el Título IX, artículos 101 a 108. Esta cuestión podía haberse resuelto, como en otras disposiciones vigentes, incluyendo la adaptación de referencias en una disposición adicional.
Al crearse la Agencia Española del Medicamento (AEM) y al aprobarse posteriormente su estatuto, se le asignan las funciones de planificación, coordinación, evaluación y desarrollo del SEFV, y se adscribe el Comité de Seguridad de medicamentos de uso humano. El RD 711/2002 desarrolla las funciones de la AEM en materia de farmacovigilancia.
Estructura orgánica
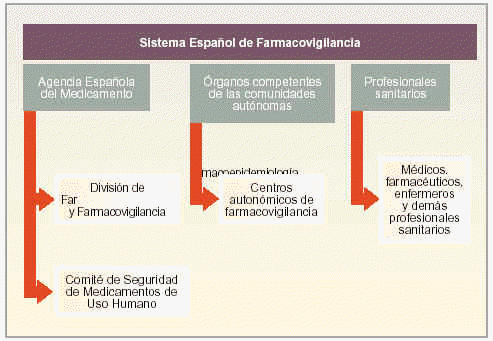
El SEFV está formado por los siguientes organismos (fig. 1):
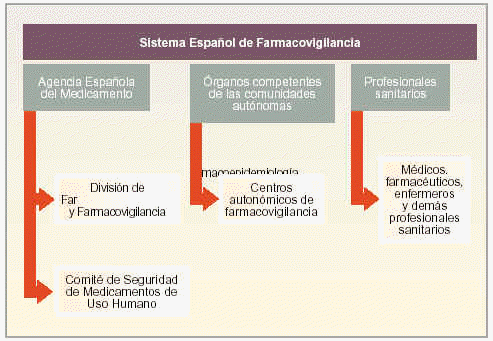
Fig. 1. Organigrama del SEFV.
La Agencia Española del Medicamento.
Los órganos competentes en materia de farmacovigilancia de las comunidades autónomas.
Los profesionales sanitarios.
Agencia Española del Medicamento
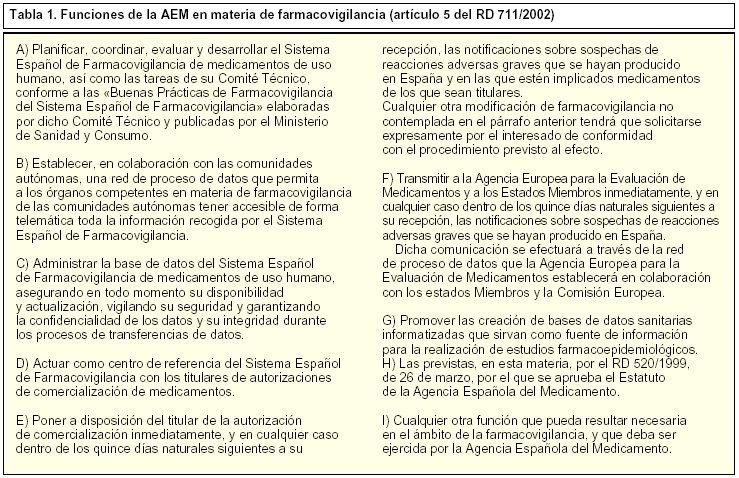
El RD 520/1999, de 26 de marzo, por el que se aprueba el Estatuto de la AEM, le atribuye, en su artículo 5.19, la obligación de planificar, coordinar, evaluar y desarrollar el SEFV. Las funciones concretas en materia de farmacovigilancia se desarrollan en el artículo 5, del Real Decreto 711/2002, de 19 de julio, cuyo contenido se transcribe en la tabla 1.
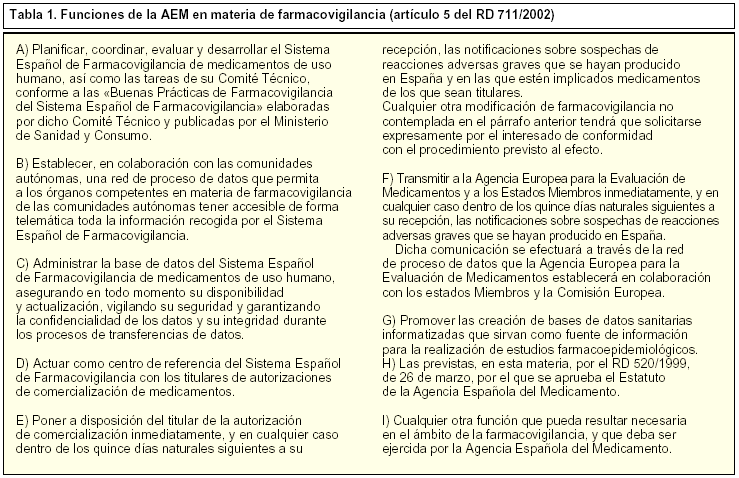
Estas funciones se desarrollan a través de dos estructuras:
La División de Farmacoepidemiología y Farmacovigilancia de la Subdirección General de Seguridad de Medicamentos.
El Comité de Seguridad de medicamentos de uso humano.
La División de Farmacoepidemiología y Farmacovigilancia es la estructura que planifica, evalúa y desarrolla el Sistema Español de Farmacovigilancia y actúa como centro nacional de referencia en materia de farmacovigilancia de medicamentos de uso humano. Este organismo se encarga de recibir, valorar, procesar y emitir la información sobre sospechas de reacciones adversas de medicamentos de uso humano2 recibida de la industria farmacéutica.
El Comité de Seguridad de medicamentos de uso humano asesora técnica y científicamente a la División de Farmacoepidemiología y Farmacovigilancia de la AEM en materia de efectos adversos o tóxicos de los medicamentos de uso humano. Sus funciones específicas se resumen a continuación:
Propone a la AEM la realización de los estudios y las investigaciones que estime necesarios para el mejor ejercicio de la farmacovigilancia de medicamentos de uso humano.
Asesora a la AEM en el ejercicio de la función de coordinación que a ésta compete, respecto a la planificación y el desarrollo del Sistema Español de Farmacovigilancia en lo referente al medicamento de uso humano.
Informa preceptivamente en el procedimiento de suspensión o revocación de una autorización de comercialización de medicamentos de uso humano en los supuestos previstos en la Ley del Medicamento.
Presta asesoramiento técnico a los representantes españoles en los grupos de trabajo y reuniones en materia de farmacovigilancia de medicamentos de uso humano que se celebren en la Unión Europea.
Con carácter facultativo, a petición del director de la AEM, colabora en la evaluación de los estudios de fase iv e informes periódicos de seguridad.
Los informes que emite el Comité de Seguridad de medicamentos de uso humano en ningún caso son vinculantes. Forman este comité: el director de la AEM, el subdirector general de Seguridad de Medicamentos, el subdirector general de medicamentos de uso humano, 6 vocales de las administraciones sanitarias de las comunidades autónomas designados por 4 años por el Ministerio de Sanidad y Consumo y 6 vocales de libre designación, seleccionados entre profesionales y expertos con conocimientos en farmacovigilancia, evaluación y control de medicamentos designados por 4 años por el Ministerio de Sanidad y Consumo.
Órganos competentes en materia de farmacovigilancia de las comunidades autónomas
En este modelo descentralizado para el control de reacciones adversas a medicamentos en España es fundamental la participación activa de las comunidades autónomas. En la actualidad, se hallan en funcionamiento 17 centros autonómicos de farmacovigilancia, su directorio se incluye en el sitio web de la AEM (www.agemed.es) actualizado a 21 de julio de 2002. Su participación no es homogénea entre todas las comunidades sino que depende de distintos factores como el volumen de población, la dispersión geográfica, el momento de incorporación al sistema o el número de profesionales sanitarios. Sus funciones concretas se encuentran descritas en el artículo 6 del RD 711/2002, de 19 de julio (tabla 2).

Profesionales sanitarios
El artículo 58.3 de la Ley del Medicamento dispone que todo profesional sanitario está obligado a notificar a las autoridades sanitarias las sospechas de reacciones adversas a medicamentos de las que tengan conocimiento y a colaborar con el SEFV. Esto significa que todos los profesionales sanitarios, es decir, médicos, odontólogos-dentistas, farmacéuticos y ATS/DUE deben participar en el sistema.
Las obligaciones concretas encomendadas a los profesionales sanitarios se desarrollan en el artículo 7 del nuevo Real Decreto (tabla 3).

Funcionamiento del SEFV
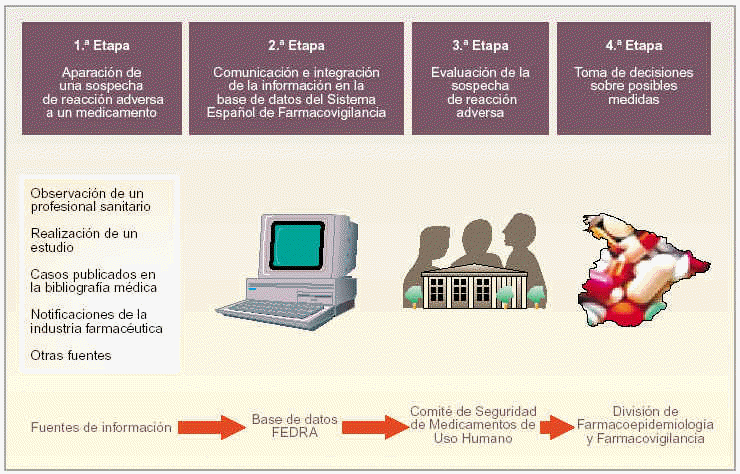
Para mejorar la comprensión de funcionamiento del SEFV, dividimos el proceso desde la aparición de una reacción adversa hasta la adopción, en caso necesario, de una medida reguladora en cuatro etapas (fig. 2):
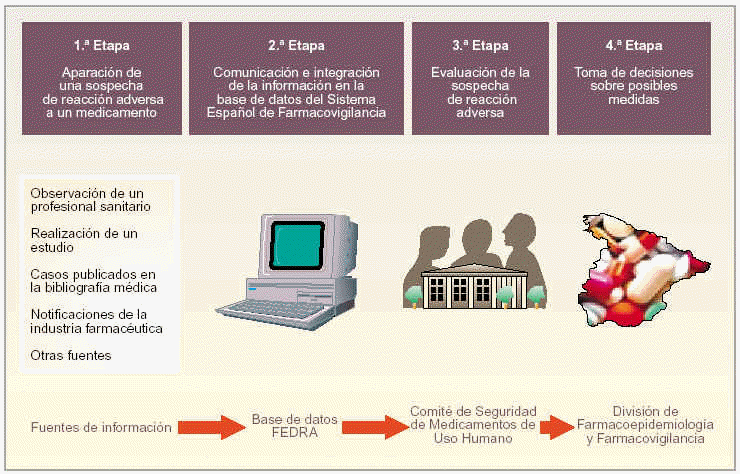
Fig. 2. Funcionamiento del SEFV.
Primera etapa. Aparición de una sospecha de reacción adversa a un medicamento.
Segunda etapa. Comunicación e integración de la información en la base de datos FEDRA.
Tercera etapa. Evaluación de la sospecha de reacción adversa.
Cuarta etapa. Toma de decisiones sobre posibles medidas reguladoras
Es un sistema fácil y muy ágil de identificación de problemas de seguridad en los medicamentos.
Aparición de una sospecha de reacción adversa a un medicamento
Las sospechas de reacciones adversas a medicamentos pueden proceder de distintas fuentes. En el artículo 3 del RD 711/2002, de 19 de julio, se especifican las fuentes válidas de obtención de información para el SEFV.
El SEFV puede recibir notificaciones de distintas procedencias aunque la fuente original de la información sea la misma.
Todo profesional sanitario puede observar durante la realización de su trabajo una sospecha de reacción adversa a un medicamento. En este momento, puede proceder a su notificación al centro autonómico de farmacovigilancia de su correspondiente comunidad autónoma mediante un formulario estandarizado que es la tarjeta amarilla3. Los distintos centros autonómicos son los encargados de recoger las notificaciones de reacciones adversas a medicamentos.
La realización de un estudio efectuado por un centro autonómico (por ejemplo, un estudio epidemiológico) o por un laboratorio farmacéutico (por ejemplo, un estudio preclínico de experimentación animal, un estudio clínico de un medicamento o bien un estudio realizado posteriormente a la autorización de un medicamento) también puede proporcionar información sobre sospecha de reacciones adversas a medicamentos.
Los casos de reacciones adversas a medicamentos publicados en la bibliografía médica, en revistas nacionales que son seguidas por los centros autonómicos o bien en revistas extranjeras seguidas a través de bases de datos informatizadas también son una posible fuente de información.
Además, la industria farmacéutica recibe de sus especialidades farmacéuticas notificación de sospecha de reacciones adversas y las comunica directamente a la División de Farmacoepidemiología y Farmacovigilancia de la AEM, quien actúa de Centro Coordinador del SEFV.
Otras fuentes de información de reacciones adversas pueden ser las autoridades sanitarias u organismos sanitarios internacionales.
Comunicación e integración de la información en la base de datos del SEFV
Los datos correspondientes a las distintas notificaciones los analizan y evalúan los distintos centros de farmacovigilancia. A la recepción de la notificación, el centro envía una carta de agradecimiento a la fuente de información, le ofrece consulta terapéutica y, si lo requiere, le solicita información adicional. Después del análisis y la evaluación, el centro procede a la codificación y el almacenamiento de la sospecha de reacción adversa en la base de datos FEDRA (Farmacovigilancia Española Datos de Reacciones Adversas).
FEDRA consiste en una base de datos única, descentralizada porque cada centro de farmacovigilancia autonómico es usuario y al mismo tiempo generador de información sobre reacciones adversas a medicamentos e integrada con el resto de las bases de datos del Ministerio de Sanidad y Consumo. Cada centro autonómico está conectado en tiempo real con la base de datos.
Este proceso se desarrolla de forma que la confidencialidad de los datos, tanto del paciente como del notificador, quede completamente garantizada.
Evaluación de la sospecha de reacción adversa
Las notificaciones de reacciones adversas son en principio sospechas y no reacciones adversas consumadas. Por ello, a partir de las notificaciones espontáneas recibidas e incorporadas en el Sistema FEDRA, la División de Farmacoepidemiología y Farmacovigilancia y el Comité de Seguridad de medicamentos de uso humano de la Agencia Española del Medicamento deben evaluar la causalidad, es decir, la relación directa entre la toma del medicamento y la aparición de la reacción adversa.
Esta evaluación conduce a la decisión por parte de la División de Farmacoepidemiología y Farmacovigilancia sobre la necesidad de tomar alguna acción reguladora.
Toma de decisiones sobre posibles medidas reguladoras
En el caso de existir la necesidad de tomar una acción reguladora, estará en relación con el tipo de reacción y con su gravedad. Pueden darse diferentes situaciones como:
El medicamento puede seguir en el mercado, pero se modifican las condiciones de uso que se autorizaron (por ejemplo: limitación de las indicaciones terapéuticas, inclusión de nuevas advertencias o precauciones en la ficha técnica y prospecto, modificación de la presentación del medicamento).
El medicamento puede seguir en el mercado, pero se distribuye la información de seguridad a todos los profesionales sanitarios.
El medicamento puede seguir en el mercado, pero se obliga a la realización de estudios específicos por parte de la compañía farmacéutica.
El medicamento no puede seguir en el mercado y se retira.
Otras actividades del SEFV
Los centros de farmacovigilancia, en paralelo a la recepción y evaluación de notificación de reacciones adversas a medicamentos, desarrollan otras actividades relacionadas con la farmacovigilancia.
Los centros de farmacovigilancia realizan reuniones con los posibles notificadores para informarles sobre problemas generales de seguridad de los medicamentos y tratan de estimularles a la notificación. A su vez, son responsables de la edición y distribución de las tarjetas amarillas a los profesionales sanitarios de su comunidad autónoma.
Los centros de farmacovigilancia publican boletines periódicos de información sobre reacciones adversas y acciones reguladoras tomadas.
Por último, cabe añadir que los centros de farmacovigilancia responden a consultas terapéuticas recibidas en su centro.
A pesar de los importantes y continuos avances en materia de farmacovigilancia, ésta sigue siendo una disciplina objeto de estudio y mejora
Consideración final
A pesar de los importantes y continuos avances en materia de farmacovigilancia, ésta sigue siendo una disciplina objeto de estudio y mejora. Buena prueba de ello es la propuesta, que se encuentra en elaboración, de modificación del Código Comunitario Relativo a los medicamentos de uso humano. Esta propuesta incorpora, entre otras, modificaciones sustanciales en materia de farmacovigilancia con objeto de potenciar el concepto de la prevención. En concreto nos referimos a:
Aumento de la frecuencia de presentación de informes periódicos en materia de seguridad a las autoridades sanitarias.
En medidas de urgencia, se habilita a la comisión para que pueda solicitar a los estados miembros la adopción de medidas temporales con efecto inmediato.
Refuerzo de las inspecciones relacionadas con las obligaciones de las compañías farmacéuticas titulares.
Mejora de la coordinación entre los estados miembros. Esta mejora se traduce en una buena comunicación entre estados en referencia a medicamentos autorizados por procedimiento de reconocimiento mutuo, obligación de utilizar una terminología común para la redacción de informes sobre reacciones adversas (terminología MedDRA) y armonización de los sistemas de farmacovigilancia de los distintos estados miembros.
En conclusión, la farmacovigilancia se encuentra en pleno proceso evolutivo y se espera de ella que nos proporcione al fin la certeza de que los medicamentos comercializados son seguros.
Notas
1. RD 711/2002, de 19 de julio, por el que se regula la Farmacovigilancia de medicamentos de uso humano (BOE de 20 de julio de 2002). En su Disposición derogatoria única, deroga los artículos 37 y 37 bis del RD 767/1993, de 21 de mayo, en su redacción dada por el RD 2000/1995, de 7 de diciembre, por el que se modifica el RD 767/1993, de 21 de mayo, que regula la evaluación, autorización, registro y condiciones de dispensación de especialidades farmacéuticas y otros medicamentos de uso humano fabricados industrialmente.
2. Según el artículo 2 c) del RD 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, por reacción adversa cabe entender «cualquier respuesta a un medicamento que sea nociva y no intencionada, y que tenga lugar a dosis que se apliquen normalmente en el ser humano para profilaxis, el diagnóstico o el tratamiento de enfermedades, o para la restauración, corrección o modificación de funciones fisiológicas».
3. Según el artículo 2 j) del RD 711/2002, de 19 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano, por tarjeta amarilla cabe entender el «formulario para la notificación de sospechas de reacciones adversas, distribuida por los órganos competentes en materia de farmacovigilancia de las comunidades autónomas a los profesionales sanitarios».
