


LA DREPANOCITOSIS es un grupo de trastornos eritrocitarios hereditarios que provoca la producción anormal de hemoglobina, el componente de los glóbulos rojos que transporta el oxígeno a los tejidos. La drepanocitosis es una enfermedad crónica que puede provocar muchas complicaciones sistémicas, como disfunción de órganos e infección causada por una perfusión tisular y una oxigenación inadecuadas.
Este artículo presenta una visión general de los síntomas y signos, complicaciones y opciones de tratamiento actuales.
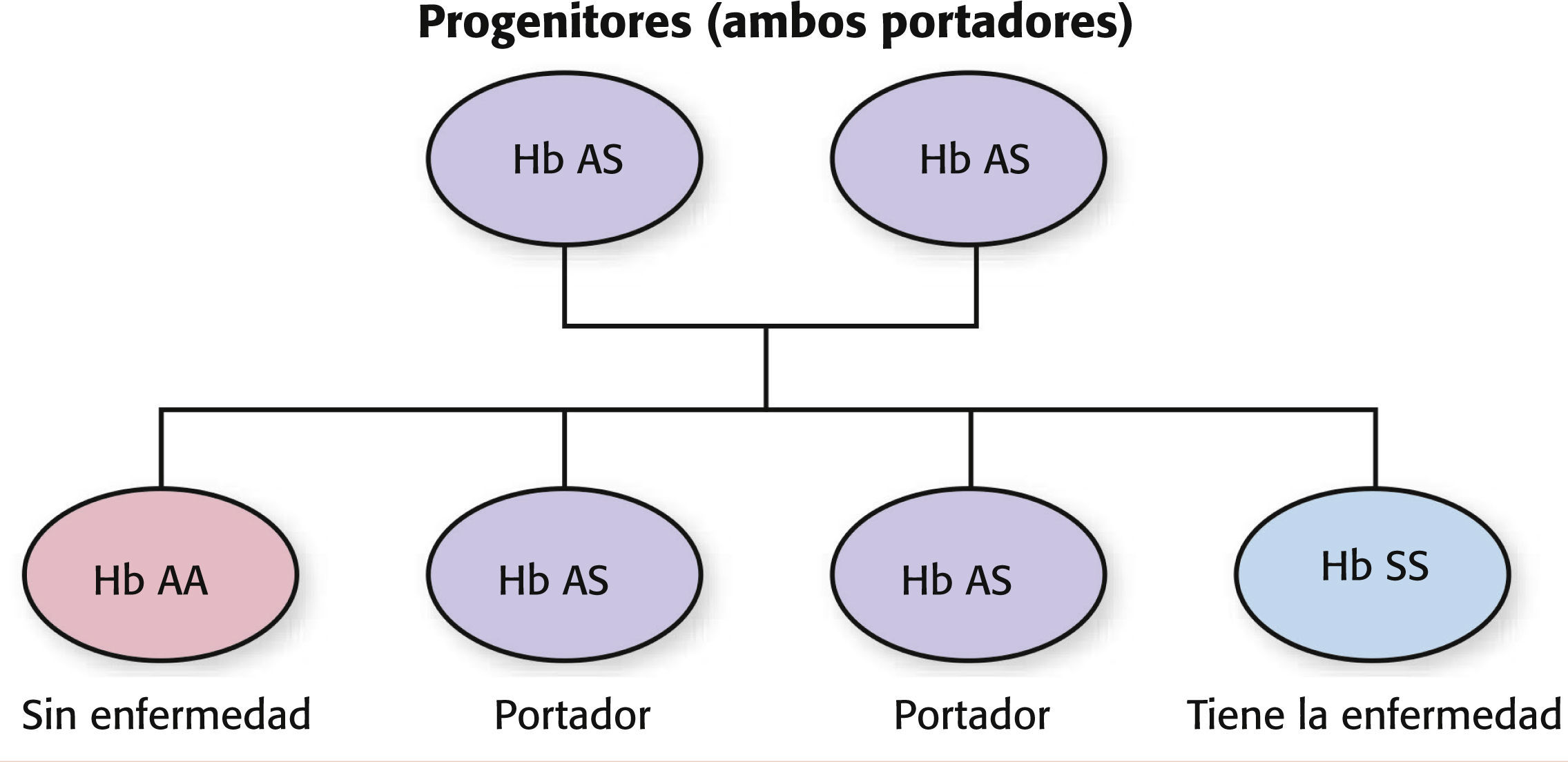
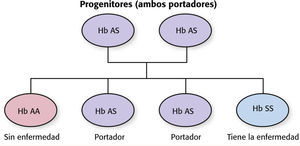
Drepanocitosis frente a rasgo drepanocíticoEs importante distinguir entre drepanocitosis y rasgo drepanocítico. En todas las formas de drepanocitosis, la persona afectada ha heredado de un progenitor, al menos, un gen de la forma anómala de la hemoglobina “S” (HbS) de células falciformes más otro gen anómalo de la hemoglobina del otro progenitor. En la forma más frecuente y grave de drepanocitosis, la anemia drepanocítica de tipo HbSS, la persona ha heredado dos genes de células falciformes (“S”), uno de cada progenitor. En otros tipos de drepanocitosis, que se diferencian en incidencia y gravedad, además del gen de la forma anómala de la hemoglobina “S” de células falciformes, la persona ha heredado otro gen anómalo (C, D, E, O, talasemia beta0 [β0] o talasemia beta+ [β+])1.
A diferencia de aquellas personas con drepanocitosis, que han heredado dos genes de hemoglobina anómalos, las personas con rasgo drepanocítico han heredado un gen de la hemoglobina “S” de células falciformes de uno de los progenitores más un gen normal de la hemoglobina “A” del otro progenitor (v. el cuadro Comprender la herencia de la drepanocitosis). A la persona con drepanocitosis se la designa como tipo HbSA. El individuo con drepanocitosis rara vez tiene problemas de salud relacionados con las células falciformes y puede esperar vivir una vida normal, pero puede transmitir la drepanocitosis a su descendencia. Esta es una diferencia importante2.
PrevalenciaLa drepanocitosis es la enfermedad eritrocitaria hereditaria más frecuente en Estados Unidos3. Durante décadas, la drepanocitosis fue considerada una enfermedad infantil porque pocos pacientes llegaban a la edad adulta. Sin embargo, en 1980, el 50% de los niños con drepanocitosis sobrevivía hasta los 20 años. Hoy día, la esperanza de vida media oscila entre 40 y 60 años4.
La mayoría de los afectados es de ascendencia africana; una minoría procede de Centroamérica o Sudamérica, de Oriente Medio, del Mediterráneo (de ascendencia turca, italiana o griega) o de la India5. Según los Centers for Disease Control and Prevention (CDC), aproximadamente 1 de cada 365 afroamericanos nacidos vivos y 1 de cada 16 300 hispanos nacidos vivos sufre drepanocitosis y esta afecta a 100 000 estadounidenses4.
En Estados Unidos se lleva a cabo una detección sistemática de drepanocitosis en recién nacidos. La amniocentesis realizada para obtener un diagnóstico prenatal puede utilizarse para detectar el gen de la célula falciforme en el líquido amniótico6 (v. el cuadro Drepanocitosis en cifras).
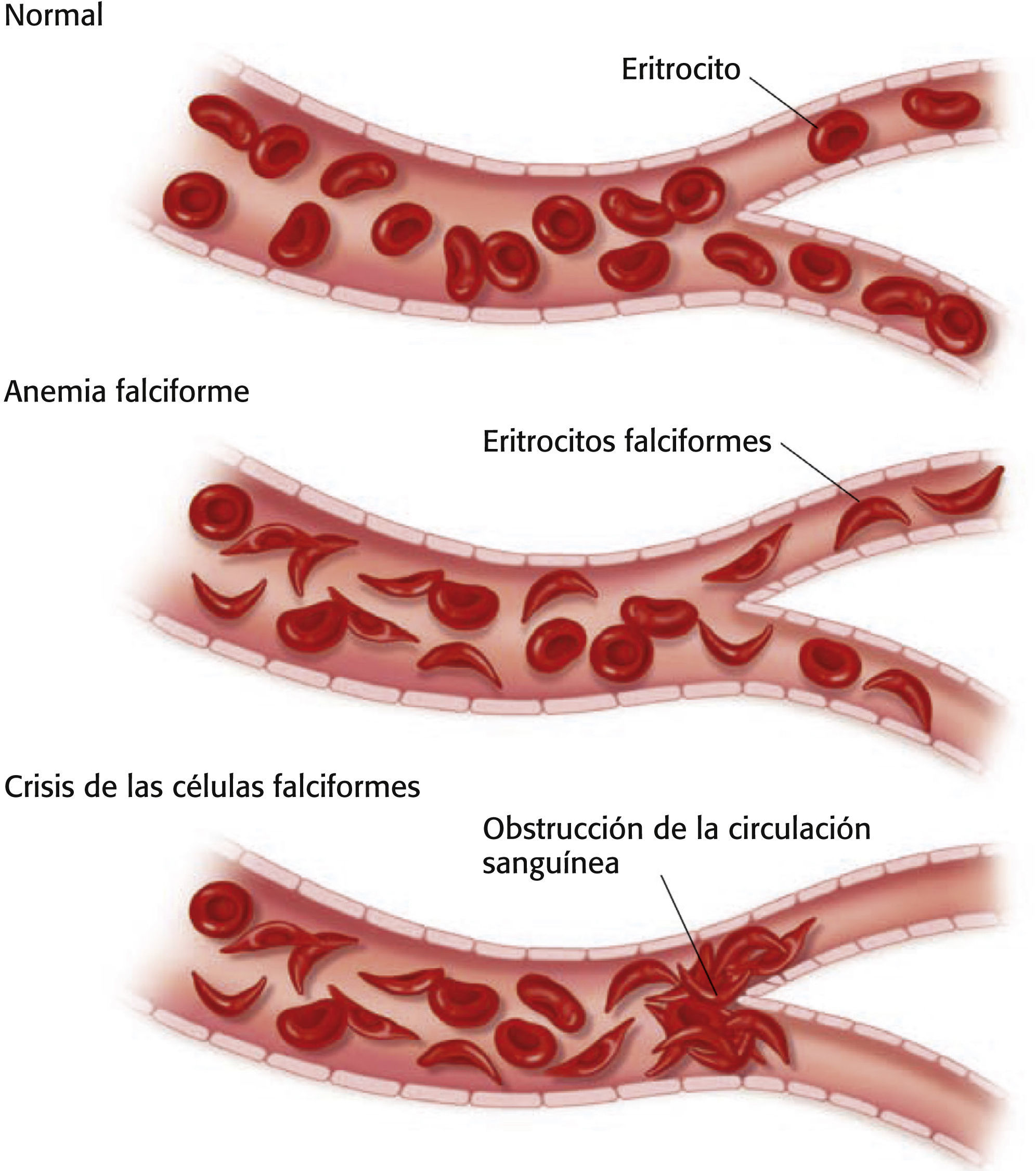
FisiopatologíaLa hemoglobina anómala de células falciformes de la drepanocitosis es menos soluble que la hemoglobina fetal o adulta normal7,8. Cuando el nivel de oxígeno en sangre y el pH de esta son bajos, se producen cambios intracelulares y la HbS adopta una formación similar al cristal, por la cual los eritrocitos pierden su forma de disco redonda, flexible y bicóncava, y se deforman, se vuelven rígidos y adoptan una forma de hoz o media luna8. Los eritrocitos falciformes tienen mayor capacidad de adherencia, disminuye su capacidad para cambiar de forma cuando se mueven por la circulación (deformabilidad) y aumenta la permeabilidad de la membrana eritrocitaria8–12.
La viscosidad de la sangre también aumenta debido a una combinación de propiedades intrínsecas e interacciones anómalas de eritrocitos falciformes con leucocitos, plaquetas, endotelio vascular y factores de coagulación. Debido a la falta de deformabilidad y la adherencia mejorada de los eritrocitos falciformes, estos se agrupan entre sí, se adhieren al endotelio vascular y obstruyen la luz del vaso8.
El proceso de formación de eritrocitos falciformes se produce a lo largo del tiempo; si el eritrocito se expone a cantidades adecuadas de oxígeno antes de que la membrana se vuelva demasiado rígida, puede volver a una forma normal. De no ser así, la aglutinación de los eritrocitos falciformes provoca una oclusión vascular y una disminución de la perfusión tisular, lo que origina complicaciones como dolor, isquemia orgánica específica e infarto13 (v. el cuadro Figura sobre la crisis de las células falciformes).
Las células falciformes también son frágiles y fuerzas mecánicas las destruyen rápidamente cuando circulan. Mientras que los eritrocitos normales viven de 90 a 120 días, la vida de una célula falciforme dura solo de 10 a 20 días. La destrucción de los eritrocitos es más rápida que el tiempo que necesita la médula ósea para reemplazarlos, lo que provoca anemia hemolítica y compromete gravemente su capacidad de transportar el oxígeno de la sangre13.
Las principales manifestaciones de la drepanocitosis son anemia hemolítica crónica y crisis vasooclusiva aguda (CVOA), que se caracterizan principalmente por lesión tisular de isquemia-reperfusión14,15. La drepanocitosis se caracteriza por la vasooclusión aguda y crónica que puede producir episodios dolorosos agudos, síndrome torácico agudo, lesión de órganos, accidente cerebrovascular e, incluso, la muerte13.
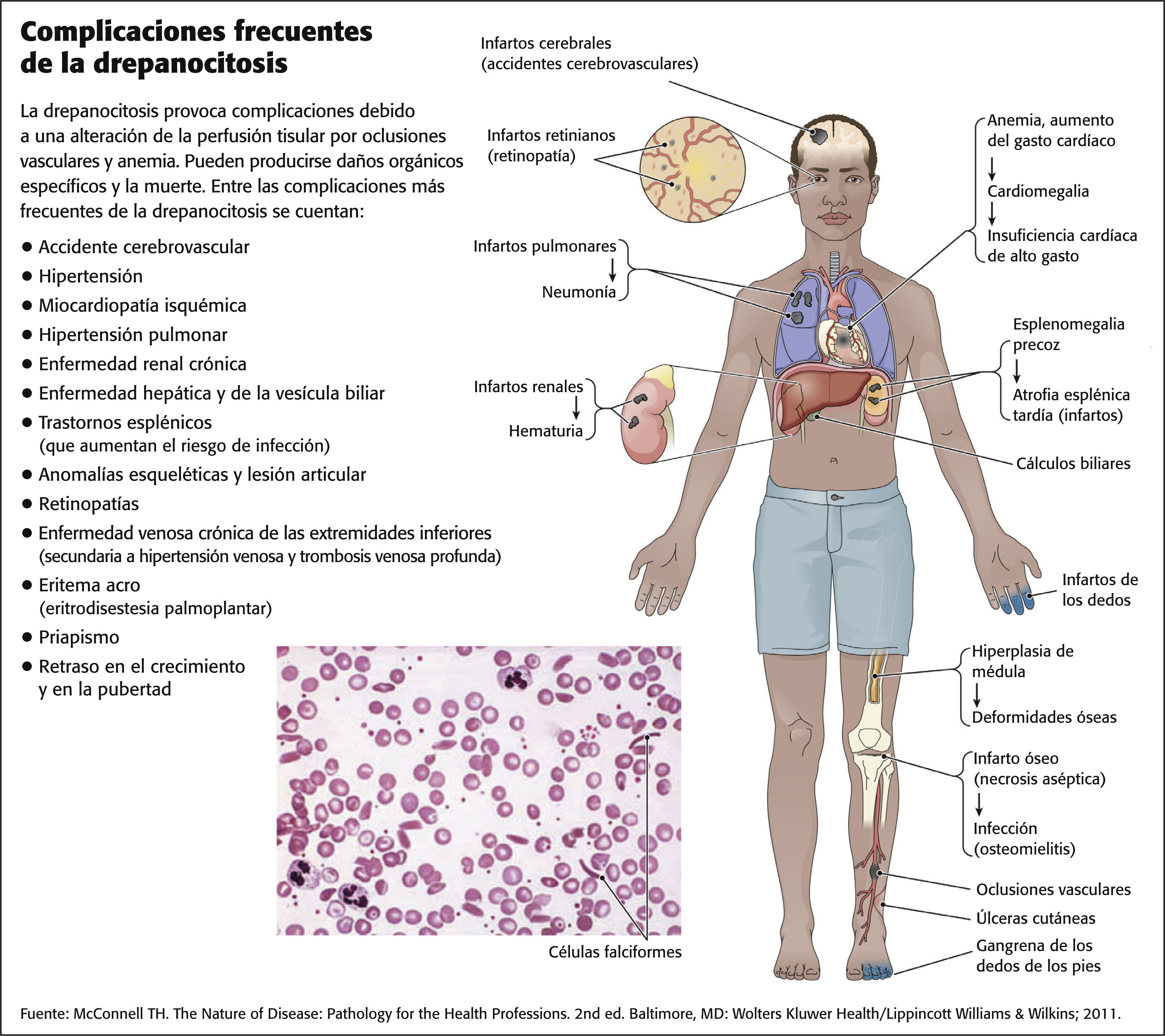
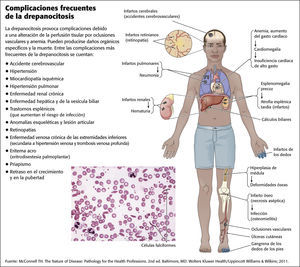
Vasooclusión en la drepanocitosisLos eritrocitos se adhieren a las paredes de los vasos sanguíneos y provocan macro y microvasooclusión (VO). La VO, junto con la anemia hemolítica, produce hipoxia, acidosis, inflamación, isquemia e infarto tisular. La hipoxia incrementa aún más la formación de eritrocitos falciformes, a la vez que empeora y perpetúa la VO. El estado protrombótico que se produce a partir de la activación de la trombina, la disminución de los niveles de anticoagulantes circulantes, la alteración de la fibrinólisis y la activación plaquetaria también están implicados en la patogenia de la VO16. Como resultado de la VO, los pacientes corren un riesgo considerable de lesión orgánica específica y otras complicaciones. Las complicaciones cardiovasculares, pulmonares y otras complicaciones relacionadas con la VO son causas principales de morbilidad y mortalidad para todos los individuos con drepanocitosis9,17 (v. el cuadro Complicaciones frecuentes de la drepanocitosis).
La Organización Mundial de la Salud considera que más de 310 000 personas nacen cada año con drepanocitosis; la mayoría de ellas, en África subsahariana41. La incidencia prevista de drepanocitosis es de 1 de cada 625 nacimientos. Sin embargo, debido al aumento de la emigración, la drepanocitosis es cada vez más frecuente en todo el mundo. Aproximadamente, de 25 a 30 millones de personas en todo el mundo tienen drepanocitosis y cerca de 100 000 viven en Estados Unidos8,9,39,42.
La VO también produce episodios de dolor agudo, denominados crisis vasooclusivas (CVO)18. La CVO es la causa más frecuente de hospitalizaciones de los pacientes con drepanocitosis. El dolor relacionado con la CVO está provocado por mediadores inflamatorios liberados por eritrocitos, macrófagos, mastocitos y plaquetas dañados, que activan los receptores nocisensibles del dolor19–21.
Entre las situaciones que pueden desencadenar una CVO se pueden citar infección, embarazo, consumo de alcohol o tabaco, exposición a temperaturas extremas, deshidratación, exposición a gran altitud, esfuerzo físico extremo y estrés; sin embargo, a menudo no se puede identificar un desencadenante19–21.
El dolor es repentino y generalmente se manifiesta en la parte inferior de la espalda, en una articulación o más, y en las extremidades. El dolor puede ser localizado o migratorio, y se describe normalmente como continuo y pulsátil. Debido al dolor, los pacientes pueden gruñir, gemir, llorar, retorcerse, doblarse y adoptar posturas raras en un intento por obtener alivio. La combinación de hipoxia, lesión por reperfusión, inflamación y lesión isquémica tisular provoca que la experiencia de dolor relacionada con una CVO de la drepanocitosis sea única19–21.
Los episodios de CVO pueden producirse desde el primer año de vida en adelante y provocan daño y lesión tisular y orgánica permanente. Debido a la naturaleza crónica de la drepanocitosis, la gravedad y la frecuencia de las crisis de dolor tienen periodos de mejoría y recaídas, y pueden repetirse de forma impredecible. Cada caso de crisis dolorosa se relaciona con lesión inflamatoria residual que se acumula y puede provocar disfunción orgánica e, incluso, insuficiencia orgánica19. El dolor que provoca la CVO puede durar horas o días. El número de crisis de dolor por año varía ampliamente, pues algunos pacientes experimentan un dolor constante de escasa intensidad.
TratamientoLos objetivos del tratamiento de la drepanocitosis incluyen aumentar al máximo la oxigenación/perfusión de los tejidos, prevenir y tratar las complicaciones relacionadas con la anemia y la VO, prevenir y tratar activamente la infección y controlar el dolor8,22. La drepanocitosis es una enfermedad crónica.
Hidroxiurea oral. La administración oral a largo plazo de hidroxiurea reduce o previene muchas complicaciones agudas y crónicas de la drepanocitosis23. La hidroxiurea se absorbe rápidamente, tiene una biodisponibilidad casi completa y es terapéutica con dosificación oral de una vez al día8. Décadas de investigación han establecido que el tratamiento más eficaz de la drepanocitosis es aumentar la hemoglobina circulante normal o fetal (HbF) y reducir la HbSS. El aumento de la concentración de HbF es el efecto principal de la hidroxiurea. Entre otros beneficios de la hidroxiurea se cuentan reducir el número de leucocitos circulantes y reticulocitos, y disminuir su adhesividad, lo que puede reducir la oclusión vascular que provoca CVO. La hidroxiurea también aumenta el tamaño de los eritrocitos y mejora la deformabilidad celular, lo que aumenta la circulación sanguínea y reduce la VO. El óxido nítrico, un vasodilatador, se libera directamente desde el metabolismo de la hidroxiurea y puede contribuir a la vasodilatación local. El tratamiento con hidroxiurea reduce considerablemente la frecuencia de episodios dolorosos relacionados con CVO y la necesidad de transfusiones y hospitalizaciones17,24,25.
Los eritrocitos falciformes (en forma de hoz o media luna) indican anemia de células falciformes. La crisis de las células falciformes provoca obstrucción de la circulación sanguínea y dolor.
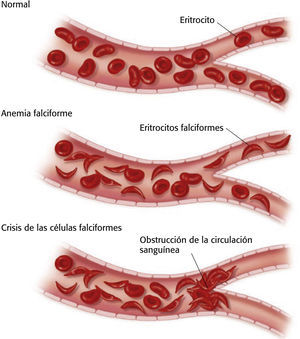
Fuente: Creason C. Stedman's Medical Terminology. 2nd ed. Baltimore, MD: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011.
Aunque la hidroxiurea no es tan eficaz como el tratamiento de transfusión sanguínea en la prevención del accidente cerebrovascular o la CVO, es el tratamiento más ampliamente utilizado y ha demostrado su eficacia en niños y adultos con drepanocitosis. La hidroxiurea es el único tratamiento farmacológico aprobado por la Food and Drug Administration para la inducción de HbF en adultos con drepanocitosis y también está aprobado por la Agencia Europea de Medicamentos para niños y adultos con drepanocitosis. El beneficio está directamente relacionado con la cantidad de HbF producida en respuesta al fármaco. Sin embargo, varios estudios clínicos han demostrado que las respuestas individuales al tratamiento con hidroxiurea son diferentes. Los niveles de HbF inducidos oscilan entre el 10% y más del 30% incluso entre los pacientes con regímenes de dosificación similares8.
El tratamiento con hidroxiurea está indicado para niños a partir de 5 años que presentan complicaciones graves de la drepanocitosis. Se recomienda incluso para niños de 9 meses, incluso para aquellos sin síntomas y para niños de tan solo 18 meses con dolor frecuente, anemia sintomática grave o episodios vasooclusivos graves. Debe realizarse un hemograma completo con regularidad en pacientes en tratamiento con hidroxiurea para detectar si existen pruebas de inmunosupresión, como la leucopenia8,23. Debe realizarse un hemograma completo, por lo menos, cada 4 semanas cuando se ajuste la dosis del paciente8.
No se conocen efectos adversos y problemas de seguridad relacionados con el uso prolongado de la hidroxiurea, pero los pacientes con drepanocitosis que tienen niveles más altos de HbF circulante tienen menos dolor, menor morbilidad y una supervivencia mejorada25,26.
Para interrumpir la CVO en su etapa prodrómica, se puede establecer un ensayo de hidratación, antiinflamatorios, analgésicos y, posiblemente, vasodilatadores. El tratamiento para pacientes que experimentan CVO incluye la terapia para interrumpir el episodio de dolor y limitar las lesiones tisular y orgánica. Las intervenciones para evitar el dolor incluyen fármacos antiinflamatorios no esteroideos para el dolor de leve a moderado si no están contraindicados y opioides como la morfina para un dolor más intenso. El tratamiento rápido y agresivo del dolor agudo puede ayudar a prevenir el dolor crónico y mejorar la calidad de vida. El tratamiento debe basarse en la clasificación de intensidad del dolor del paciente y en lo que se haya utilizado con éxito en el pasado8,18.
Tratamiento de transfusión sanguínea. La decisión de transfundir sangre debe basarse en una evaluación de riesgo y beneficio, generalmente después de consultar a un experto, como un hematólogo8. Puesto que los eritrocitos falciformes promueven la VO, la sangre normal del donante se transfunde para suprimir ese proceso. La transfusión sanguínea reemplaza la HbS anómala con hemoglobina normal (HbA) para interrumpir una CVO al aumentar la perfusión y, posteriormente, el suministro de oxígeno celular3,17,27. La transfusión de sangre cada 3-4 semanas incrementa la concentración de HbA, reduce la HbSS, lo que mejora el flujo sanguíneo con eritrocitos no falciformes y suprime la producción de eritrocitos falciformes. No se conoce el nivel óptimo para elevar la HbA con una transfusión sanguínea, pero un objetivo terapéutico efectivo para la terapia transfusional es mantener la HbS en menos del 30% de la hemoglobina total28.
El tratamiento de transfusión sanguínea requiere la formación del paciente, la concienciación sobre beneficios y riesgos, el consentimiento informado, el establecimiento de acceso vascular adecuado y apropiado, y la preparación y administración de múltiples unidades.
Terapias en investigación. En la actualidad, la única cura para la drepanocitosis es un trasplante de células hematopoyéticas (TCH). El TCH es un tratamiento en evolución con muchas complicaciones y se limita a los niños de 16 años o menores con síntomas y signos graves que no responden a la hidroxiurea y el tratamiento de transfusión, y que tienen un donante hermano emparejado29. Las investigaciones en curso que utilizan la terapia génica para introducir genes correctores en la médula ósea también son prometedoras30.
ComplicacionesLas transfusiones sanguíneas se asocian con varios riesgos posibles, entre los cuales se cuentan hiperviscosidad, aloinmunización, hemólisis y sobrecarga de hierro. Si se administran transfusiones de forma esporádica o con regularidad, introducen una carga considerable de hierro en el receptor. Puesto que las transfusiones insertan el hierro fuera de las vías normales de regulación del hierro, ello puede sobrecargar la capacidad fisiológica normal para procesar el hierro y provocar sobrecarga de hierro y la consiguiente disfunción de órganos. Aunque la sobrecarga de hierro relacionada con el número de transfusiones y el método de transfusión es posible, se puede manejar con el tratamiento de quelación de hierro8,10,31.
Entre otros riesgos potenciales relacionados con la transfusión sanguínea pueden citarse aumento de la viscosidad debido a un mayor contenido de hemoglobina, lo que podría provocar VO y una CVO dolorosa. La reacción hemolítica tardía a la transfusión puede desarrollarse de 7 a 28 días después de las transfusiones de eritrocitos debido a incompatibilidades de grupos sanguíneos. Deben tenerse en cuenta las reacciones transfusionales en pacientes con empeoramiento de la anemia, ictericia o dolor después de la transfusión8.
- •
Prevención y tratamiento de infecciones. Los pacientes con drepanocitosis son muy vulnerables a la infección y estas tienden a ser más graves que en aquellas personas sin drepanocitosis. Esto se debe, en gran parte, a la disfunción del bazo en los microinfartos secundarios a la VO que se desarrollan en la infancia. El bazo es responsable de la filtración de la sangre y de la producción de anticuerpos, por lo que un bazo que no funciona o la asplenia aumenta el riesgo de infección. A la edad de 1 año, el 30% de los pacientes pediátricos con drepanocitosis es asplénico y a los 6 años ya lo es el 90%. El riesgo de infección en niños con drepanocitosis es 400 veces mayor si se comparan las cifras con las de la población general. Hay que proteger a estos pacientes frente a la infección y mantener las inmunizaciones actuales frente a la gripe, el neumococo y el meningococo, además de las inmunizaciones infantiles habituales recomendadas8,15,17,32.
Los niños con drepanocitosis por regla general reciben antibióticos profilácticos (normalmente, penicilina o una alternativa) por lo menos hasta los 5 años. Además de otras inmunizaciones habituales, los adultos con drepanocitosis deben recibir inmunizaciones frente a la gripe y el neumococo22.
Los antecedentes de inmunización y el estado de salud deben revisarse en cada visita al médico. Debe enseñarse a los adultos, niños y familias que la fiebre (superior a 38,5°C o 101,5 °F) es una urgencia médica que requiere sin demora una evaluación, hemocultivos y tratamiento, incluyendo antibióticos33.
Todos los pacientes con drepanocitosis deben mantener una hidratación adecuada para reducir las complicaciones de la enfermedad. Los pacientes que no pueden beber líquidos adecuados necesitarán tratamiento i.v. El estado del volumen de líquidos del paciente y la posibilidad de transfusión sanguínea determinan la selección de líquidos i.v. Puesto que los pacientes con drepanocitosis pueden ver disminuida su capacidad de excreción de sodio, las enfermeras deben vigilar los niveles de sodio y estar pendientes de la hipernatremia, especialmente en pacientes que reciben solución de cloruro sódico al 0,9%. La hipernatremia puede deshidratar los eritrocitos y aumentar la formación de eritrocitos falciformes29.
Puesto que los pacientes con drepanocitosis son hipercoagulables, los adultos hospitalizados con enfermedad médica aguda deben recibir profilaxis de la tromboembolia venosa, a menos que ello esté contraindicado29,34.
- •
Drepanocitosis y accidente cerebrovascular. Los accidentes cerebrovasculares son una terrible manifestación neurológica y causan una importante morbilidad en los pacientes con drepanocitosis8,20,24.
Los accidentes cerebrovasculares isquémicos en la drepanocitosis son más frecuentes en niños y adultos mayores, y menos frecuentes en adultos de 20 a 29 años8.
Los pacientes que sufren accidentes cerebrovasculares suelen presentar síntomas y signos, como parálisis facial, disartria o afasia, paresia de las extremidades superiores y trastornos del equilibrio y visuales, que pueden estar precedidos por ataques isquémicos transitorios. Los pacientes con infartos cerebrales asintomáticos pueden presentar signos neurológicos no focales, como retrasos en el desarrollo, poco o bajo rendimiento escolar en niños, o cambios sociales o laborales en adultos. A lo largo de su vida, debe valorarse la posibilidad de que a los pacientes con drepanocitosis se les realice una evaluación neurocognitiva formal cuando las valoraciones revelen cualquiera de estas presentaciones8.
En aquellos pacientes con un déficit neurológico que sugiere un accidente cerebrovascular isquémico evolutivo agudo puede estar indicada una transfusión sanguínea, incluyendo una exanguinotransfusión completa24. La tasa de recurrencia de accidente cerebrovascular en los primeros 2 años es de, aproximadamente, el 50% si no se inicia ningún tratamiento24. Al reducir la HbSS anómala y aumentar la HbA normal, el tratamiento de transfusión sanguínea está indicado como tratamiento para el accidente cerebrovascular isquémico evolutivo agudo y como medida preventiva para el accidente cerebrovascular isquémico cuando se utiliza con la valoración de la ecografía Doppler transcraneal (EDTC; v. el cuadro ¿Qué es el estudio STOP?)21,24,28.
El accidente cerebrovascular hemorrágico es más frecuente en pacientes con drepanocitosis que tienen entre 20 y 29 años. Debido a sus características anatomopatológicas únicas, el accidente cerebrovascular hemorrágico conlleva mayor riesgo de muerte que el accidente cerebrovascular isquémico. Los factores de riesgo del accidente cerebrovascular hemorrágico relacionado con drepanocitosis incluyen hemoglobina baja, leucocitosis, transfusión de sangre en los últimos 14 días, tratamiento con corticoesteroides y CVO. Tenga en cuenta que un accidente cerebrovascular hemorrágico en la drepanocitosis no excluye la posibilidad de que coexista con un accidente cerebrovascular isquémico evolutivo agudo o pasado24.
En la drepanocitosis, la HbS puede provocar una vasculopatía cerebral grave que cause oclusión micro y macrovascular, y posterior desarrollo de vasos colaterales alrededor del polígono arterial de Willis (PAW). El PAW está situado en la base del cerebro. La vasculopatía cerebral que afecta al PAW provoca una enfermedad denominada síndrome de moyamoya, un término japonés que significa “bocanada de humo”. Se utiliza para describir el aspecto angiográfico de los vasos colaterales que surgen del PAW debido a las oclusiones que implican a estos vasos. Aunque el síndrome de moyamoya se presenta en personas sin drepanocitosis, algunos estudios han demostrado que la existencia de este síndrome aumenta considerablemente el riesgo de accidente cerebrovascular hemorrágico, sobre todo en aquellas personas con drepanocitosis. Aunque son posibles los accidentes cerebrovasculares isquémicos y hemorrágicos, la hemorragia intracraneal es más frecuente en adultos con drepanocitosis y síndrome de moyamoya35,36.
El Stroke Prevention Trial in Sickle Cell Anemia (STOP o ensayo de prevención de accidentes cerebrovasculares en la anemia de células falciformes) fue un avance importante en la detección del accidente cerebrovascular en pacientes con drepanocitosis. STOP demostró la eficacia del cribado mediante ecografía Doppler transcraneal (EDTC) para identificar a niños con alto riesgo de lesión cerebral isquémica, incluyendo el accidente cerebrovascular43,44.
El ensayo clínico STOP estudió a niños con velocidades anómalas de EDTC, definidas como 200 cm/s y superiores, que se asignaron aleatoriamente a transfusión sanguínea habitual cada 3-4 semanas o cuidados estándares, lo que significaba sin transfusión o transfusiones solo episódicas. El riesgo de accidente cerebrovascular se redujo drásticamente en el grupo transfundido44,45. El tratamiento de transfusión sanguínea de pacientes con drepanocitosis que tienen velocidades anómalas de EDTC puede reducir el riesgo de accidente cerebrovascular del 10% por año a menos del 1%, lo que genera una reducción del 90% del riesgo de accidente cerebrovascular8,10,45.
La EDTC se recomienda, al menos, una vez al año en niños y el tratamiento de transfusión sanguínea debe plantearse cuando la velocidad de la circulación sanguínea cerebral alcanza o supera el umbral de 200 cm/s. Este enfoque, conocido como Protocolo STOP, ha sido aprobado universalmente como un patrón de atención para niños con drepanocitosis con edades comprendidas entre los 2 y los 16 años44,45
El ensayo STOP demostró la eficacia de las transfusiones sanguíneas periódicas a largo plazo para la prevención primaria del accidente cerebrovascular. Los investigadores de STOP no encontraron evidencia, a pesar del control regular, de que cualquier infección transmitida por transfusión se asociara con las transfusiones preventivas administradas durante el ensayo.
Los síntomas y signos del accidente cerebrovascular hemorrágico incluyen súbito dolor de cabeza intenso, náuseas, vómitos y rápido deterioro de la función neurológica, que puede evolucionar hasta llegar al coma. Los síntomas motores o sensoriales pasajeros pueden producirse de forma similar a la actividad convulsiva.
Para el manejo adicional de pacientes con accidente cerebrovascular hemorrágico se puede proceder a la interrupción de cualquier anticoagulante o antiplaquetario. Una sustancia neutralizante puede ser necesaria en algunos tipos de tratamiento de anticoagulación. La angiografía puede usarse para identificar los vasos implicados y para guiar el tratamiento. La evaluación e intervención neuroquirúrgicas pueden ser necesarias para reducir la presión intracraneal o para tratar la rotura de un aneurisma cerebral.
Intervenciones de enfermería en pacientes con drepanocitosis- •
Compruebe las constantes vitales y clasifique la amplitud de los pulsos periféricos, la existencia de dolor, el color y la temperatura de la piel, el tiempo de llenado capilar y la pérdida de la integridad cutánea.
- •
Inicie las transfusiones eritrocitarias como se indica.
- •
Valore el deterioro del intercambio gaseoso causado por la disminución de la capacidad de transporte del oxígeno de los eritrocitos falciformes. Realice una evaluación pulmonar detallada y completa que incluya los ruidos respiratorios, la frecuencia respiratoria y la oxigenación. Evalúe los hallazgos anómalos, como disnea, ortopnea, tos, disminución de la PaO2, saturación del oxígeno inferior al 90%, existencia de cianosis y sonidos anómalos de la respiración, como crepitantes y sibilancias. Compruebe si existe dolor torácico, que puede significar síndrome torácico agudo (v. el cuadro Comprender el síndrome torácico agudo). Vigile al paciente por si hay progresión de los síntomas. Inicie tratamiento con oxígeno, broncodilatadores y esteroides inhalados si es necesario.
- •
Valore el estado del paciente por si se producen alteraciones en el volumen de líquidos y de síntomas y signos de déficit de volumen de líquidos y función renal anómala (a menudo, secundaria a microangiopatía). Evalúe los síntomas y signos de deshidratación, como piel seca y membranas mucosas, disminución de la turgencia de la piel, hipotensión y taquicardia. Controle la diuresis, la densidad de la orina, los niveles de nitrógeno ureico y creatinina, y los niveles de electrolitos séricos. Inicie el tratamiento con líquidos i.v. si es necesario.
- •
Examine la piel del paciente y trate de observar si hay heridas o edemas. Inicie el cuidado de la herida si es necesario y tome las medidas necesarias para prevenir la erosión cutánea.
- •
Realice una cuidadosa evaluación del dolor, incluyendo el tipo, intensidad, ubicación, desencadenantes del dolor y evaluación continua de las medidas de alivio del dolor21,24,35,37,38.
Los pacientes deben saber cómo manejar la enfermedad crónica y prevenir las complicaciones. Entre los temas de enseñanza pueden citarse las implicaciones genéticas de la enfermedad, como planificación familiar. Debe hacerse hincapié en la importancia del seguimiento habitual por parte de los profesionales sanitarios que sean expertos en el manejo de la drepanocitosis y de realizarse las pruebas de laboratorio según lo indicado. Debe fomentarse el autocuidado óptimo, como tomar los medicamentos recetados, informar de medicamentos sin receta o de suplementos antes de tomarlos, realizar exploraciones dentales y oculares con regularidad, mantener una buena nutrición y una hidratación adecuada, evitar temperaturas extremas y el esfuerzo físico, identificar y evitar que se desencadenen CVO, así como evitar el tabaquismo y el consumo de alcohol. Con el paciente hay que desarrollar un plan individualizado para controlar el dolor en el domicilio y enseñar al paciente a comenzar el tratamiento tan pronto como podría comenzar un episodio de CVO39. Deben promoverse estrategias de prevención de infecciones, como llevar a cabo todas las inmunizaciones recomendadas, y debe evitarse el contacto con otras personas que tienen infecciones; deben tomarse todas las dosis recetadas de antibióticos, así como proteger la piel de lesiones. Debe darse información sobre intervenciones de manejo del dolor, su evaluación y planes. Se ha de aconsejar al paciente que busque atención médica inmediatamente si aparece fiebre o cualquier signo de infección (como eritema, edema o tos); dolor articular intenso u otro dolor; dolor torácico; disminución de la cantidad de orina u orina turbia, sanguinolenta u oscura; cualquier problema de visión, y cualquier cambio en el estado neurológico8.
El síndrome torácico agudo es otra complicación aguda frecuente y grave de la drepanocitosis. Clínicamente, aunque el síndrome torácico agudo puede desarrollarse en cualquier momento, por lo general se presenta como una aparición repentina de una combinación de tos, falta de aire, tirajes y crepitaciones que están acompañadas por un nuevo infiltrado pulmonar en la radiografía torácica. Los niños suelen tener fiebre y afectación del lóbulo superior o medio, pero muchos adultos permanecen afebriles y tienen enfermedad multilobulada. El síndrome torácico agudo no presenta rasgos distintivos de laboratorio, pero la concentración de hemoglobina a menudo disminuye bruscamente. Puesto que es una de las principales causas de muerte en pacientes con drepanocitosis, los pacientes con este síndrome deben recibir un diagnóstico y un tratamiento inmediatos8,46.
En los últimos 20 años, la comprensión de la fisiopatología y el manejo de la drepanocitosis ha aumentado de manera importante. En la actualidad, la drepanocitosis puede tratarse satisfactoriamente con pruebas basadas en la evidencia y pautas de tratamiento. Sin embargo, a pesar del progreso reciente, los pacientes con drepanocitosis continúan teniendo dificultades para mantener la calidad de vida y tienen una vida útil más corta de lo normal24,40. Es posible que la investigación en curso aporte mejores tratamientos y una cura potencial de la drepanocitosis. ■
En el Brigham and Women's Hospital de Boston, Massachusetts, Vincent M. Vacca, Jr., es educador de enfermera clínica y Lora Blank es RN en neurociencia ICU.
Los autores y los editores declaran no tener ningún conflicto de intereses potencial, financiero o de otro tipo relacionado con este artículo.