






El descubrimiento de autoanticuerpos específicos es un hito muy importante en el diagnóstico de las miopatías inflamatorias idiopáticas (MII), permitiendo a los nuevos consensos de clasificación incluir en los criterios diagnósticos los hallazgos serológicos junto a los clínicos e histopatológicos usados tradicionalmente.
ObjetivoOfrecer una descripción de los anticuerpos específicos de miositis (AEM) y de los anticuerpos asociados a miositis (AAM), la metodología para su determinación, su fenotipo asociado, y su utilidad en la práctica clínica habitual.
MetodologíaRevisión bibliográfica en las bases de datos PubMed-NCBI, SciELO y LILACS.
DesarrolloSe desarrollan los AEM: anti-aminoacil-ARNt sintetasas (AAS), anti-Mi-2 (complejo de desacetilasa remodelador de nucleosomas), anti-SRP (partícula de reconocimiento de señal), anti-TIF1γ (factor transcripcional intermediario 1γ), anti-NXP-2 (proteína de matriz nuclear 2), anti-MDA5 (gen asociado a la diferenciación del melanoma 5), anti-SAE (enzima activadora de SUMO), anti-HMGCR (3-hidroxi-3-metilglutaril-CoA reductasa) y anti-cN-1A (5’nucleotidasa 1A citosólica) y los AAM: anti-PM/Scl (complejo proteico de exosomas -PM/Scl75/100), anti-U1-RNP (ribonucleoproteína pequeña U1), anti-Ku (subunidad reguladora de ADN-PK) y anti-Ro/SSA (ribonucleoproteína 52/60.).
ConclusionesLas nuevas clasificaciones de MII con la inclusión de criterios serológicos, permitirán definir poblaciones de pacientes más homogéneas, lo que mejorará el poder de los ensayos clínicos para identificar los tratamientos dirigidos a los diferentes subgrupos de pacientes.
The discovery of specific autoantibodies meant a significant milestone in mainly idiopathic inflammatory myopathies (IIM) diagnosis. This granted serological findings to be taken into account along with traditional ones – as clinical and histopathological – in diagnosis criteria, resulting in a new classification consensus.
ObjectivesTo describe myositis-specific antibodies (AEM) and myositis-associated antibodies (AAM), to describe methodologies for AEM and AAM identification, to define their utility and convenience in daily clinical practice.
DevelopmentAEMs are classified as follows: anti-aminoacyl-tRNA synthetases (AAS), anti-Mi-2 (nucleosome remodeling deacetylase complex), anti-SRP (signal recognition particle), anti-TIF1γ (transcription intermediary factor 1γ), anti-NXP-2 (Nuclear Matrix Protein 2), anti-MDA5 (Melanoma Differentiation Associated Gene 5), anti-SAE (Small ubiquitin-like modifier activating-enzyme), anti-HMGCR (3-Hydroxy-3-methylglutaryl-CoA reductase) and anti-cN-1A (Cytosolic 5′-nucleotidase 1A) and MAA: anti-PM/Scl (Exosome Protein Complex PM/Scl75/100), anti-U1-RNP (U1 small nuclear RNP), anti-Ku (DNA-PK regulatory subunit), anti-Ro/SSA (Sjögren's syndrome related antigen A).
ConclusionsThe incorporation of serological criteria in IIM diagnosis criteria led to a new classification consensus, resulting in the tracing and identification of homogeneous patient populations. Such outline provides an opportunity to improve and empower clinical trials when it comes to determine treatments directed to different patient subgroups.
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de enfermedades musculares autoinmunes, adquiridas, poco frecuentes, caracterizadas por inflamación del músculo esquelético, ya sea de forma aislada o en el contexto de una afección sistémica.
Sobre la base de los síntomas musculares, lesiones cutáneas y los hallazgos histopatológicos, se identificaron diferentes subgrupos: la dermatomiositis (DM), la polimiositis (PM), la miositis por cuerpos de inclusión esporádica (MCIe) y, en los últimos años se incluyó la miopatía necrosante inmunomediada (MNI).
Estos subgrupos dominaron los criterios de clasificación de las MII hasta la fecha1.
Una limitación importante de estos criterios es que las características histopatológicas pueden superponerse entre los subgrupos y, además, algunas características como la presencia de infiltrados inflamatorios pueden no ser específicos y encontrarse en otras miopatías, como la distrofia facioescapulohumeral, distrofias de cinturas, distrofia muscular de Duchenne, disferlinopatías, miopatías metabólicas (Pompe, McArdle, lipidosis) y miopatías degenerativas (miopatías miofibrilares, miopatía por cuerpos de inclusión hereditaria). Además, el infiltrado inflamatorio en algunos pacientes con MII puede estar ausente y las características histopatológicas pueden ser inespecíficas e incluso casi normales.
De todo esto surge la necesidad de combinar características clínicas, histopatológicas y serológicas en un intento de diferenciar los distintos subgrupos. Sobre todo, teniendo en cuenta que la respuesta al tratamiento y el pronóstico varían entre los subgrupos2.
El descubrimiento de una serie de autoanticuerpos fue un hito importante en el diagnóstico de las MII y dio lugar a nuevos criterios diagnósticos para una clasificación basada en hallazgos clínicos, histopatológicos y serológicos2-4.
En la clasificación actual se reconocen los siguientes subgrupos principales de miopatías inflamatorias: la DM, la PM, la MNI, la MCIe y la miopatía de superposición (MS), incluido el síndrome antisintetasa (SAS)5,6.
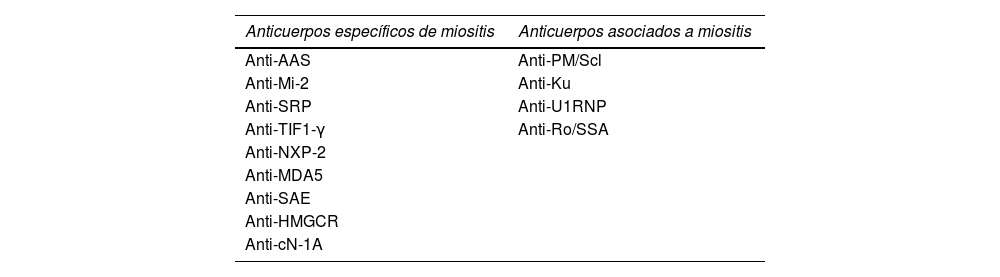
Actualmente, se pueden encontrar autoanticuerpos en hasta el 70% de los pacientes con MII y se clasifican en 2 grupos según su precisión diagnóstica: anticuerpos específicos de miositis (AEM) y los anticuerpos asociados a miositis (AAM)7 (tabla 1).
Clasificación serológica de anticuerpos en miositis
| Anticuerpos específicos de miositis | Anticuerpos asociados a miositis |
|---|---|
| Anti-AAS | Anti-PM/Scl |
| Anti-Mi-2 | Anti-Ku |
| Anti-SRP | Anti-U1RNP |
| Anti-TIF1-γ | Anti-Ro/SSA |
| Anti-NXP-2 | |
| Anti-MDA5 | |
| Anti-SAE | |
| Anti-HMGCR | |
| Anti-cN-1A |
Anti-AAS: anti-aminoacil-ARNt sintetasa; Anti-Mi2: anti-complejo de desacetilasa remodelador de nucleosomas; Anti-SRP: anti-partícula de reconocimiento de señal; Anti-TIF1-γ: anti-factor intermediario de transcripción 1 γ; Anti-NXP-2: anti-proteína de matriz nuclear 2; Anti-MDA5: anti-gen 5 asociado a la diferenciación del melanoma 5; Anti-SAE: anti-enzima activadora de SUMO-1; Anti-HMGCR: anti-3-Hidroxi-3-metilglutaril-CoA reductasa; Anti-cN-1A: anti-5’nucleotidasa 1A citosólica, Anti-PM Scl: anti-complejo proteico de exosomas (PM/Scl75/10), anti-U1 RNP: anti-ribonucleoproteína pequeña U1, anti-Ku: anti-subunidad reguladora de ADN-PK, antiRo/SSA: anti-ribonucleoproteína 52/60.
En esta revisión ofrecemos una descripción basada en la división en AEM y AAM. Se describen la metodología para su determinación, el anticuerpo con su fenotipo asociado y su utilidad en la práctica clínica habitual.
Métodos de determinaciónEn la actualidad hay múltiples técnicas disponibles para detectar anticuerpos en miositis con variable sensibilidad, especificidad, costos y complejidad, tanto para su uso clínico como en investigación. El método gold standard para la mayoría de los anticuerpos es la inmunoprecipitación (IP) de ARN con tinción de sales de plata y/o IP proteica de lisados celulares (generalmente células K562 marcadas con 35S-metionina). La IP es el método de mayor sensibilidad y especificidad, ya que evalúa la unión de los autoanticuerpos al ARN y a los complejos proteicos en su configuración nativa, pero es en método costoso, que utiliza materiales radiactivos y técnicamente complejo8. La contrainmunoelectroforesis (CIEF) y la inmunodifusión (ID) fueron los primeros métodos para detectar AEM y AAM, pero son semicuantitativos y poco sensibles, por lo que fueron sustituidos en gran medida por ensayos inmunoenzimáticos. Las ventajas del ensayo inmunoenzimático ELISA (ensayo inmunoabsorbente ligado a enzimas) son la estandarización, la reproducibilidad a gran escala y los resultados cuantitativos. Los ensayos de inmunoblot (IB) pueden detectar muchos autoanticuerpos simultáneamente y actualmente se dispone de inmunoblot multiplex como IB de puntos o de transferencia de línea (LIA)9. La sensibilidad de esta técnica varía dependiendo de la naturaleza del antígeno y no siempre provee una completa precisión en términos de especificidad y los falsos positivos son comunes10. El uso de IB para el diagnóstico de MII va en aumento, por lo que se plantea la necesidad de mejorar la estandarización, evaluación y validación de la técnica de cada laboratorio.
Autoanticuerpos específicos de miositis (tabla 2 y fig. 1)Como su nombre lo indica, los AEM son específicos para miositis autoinmune y su especificidad diagnóstica excede el 90%. Se dirigen contra ribonucleoproteínas citoplasmáticas y nucleares involucradas en procesos claves de la biología celular como la transcripción genética, síntesis y translocación de proteínas y la respuesta inmune antiviral innata10.
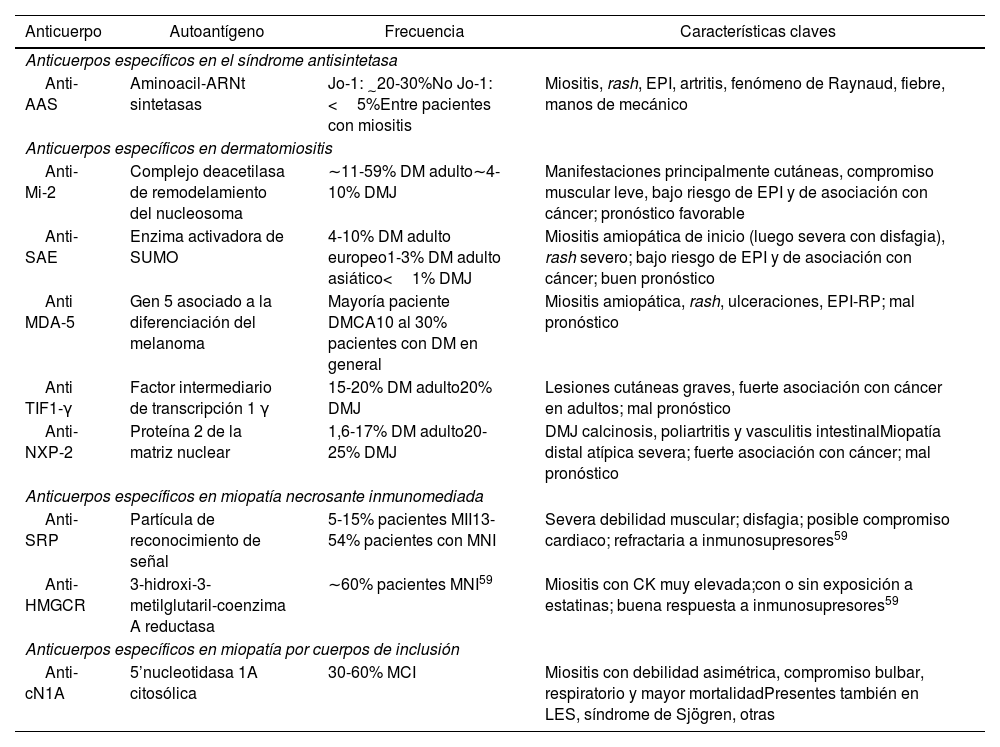
Asociaciones clínicas y frecuencia de AEM
| Anticuerpo | Autoantígeno | Frecuencia | Características claves |
|---|---|---|---|
| Anticuerpos específicos en el síndrome antisintetasa | |||
| Anti-AAS | Aminoacil-ARNt sintetasas | Jo-1: ∼20-30%No Jo-1: <5%Entre pacientes con miositis | Miositis, rash, EPI, artritis, fenómeno de Raynaud, fiebre, manos de mecánico |
| Anticuerpos específicos en dermatomiositis | |||
| Anti-Mi-2 | Complejo deacetilasa de remodelamiento del nucleosoma | ∼11-59% DM adulto∼4-10% DMJ | Manifestaciones principalmente cutáneas, compromiso muscular leve, bajo riesgo de EPI y de asociación con cáncer; pronóstico favorable |
| Anti-SAE | Enzima activadora de SUMO | 4-10% DM adulto europeo1-3% DM adulto asiático<1% DMJ | Miositis amiopática de inicio (luego severa con disfagia), rash severo; bajo riesgo de EPI y de asociación con cáncer; buen pronóstico |
| Anti MDA-5 | Gen 5 asociado a la diferenciación del melanoma | Mayoría paciente DMCA10 al 30% pacientes con DM en general | Miositis amiopática, rash, ulceraciones, EPI-RP; mal pronóstico |
| Anti TIF1-γ | Factor intermediario de transcripción 1 γ | 15-20% DM adulto20% DMJ | Lesiones cutáneas graves, fuerte asociación con cáncer en adultos; mal pronóstico |
| Anti-NXP-2 | Proteína 2 de la matriz nuclear | 1,6-17% DM adulto20-25% DMJ | DMJ calcinosis, poliartritis y vasculitis intestinalMiopatía distal atípica severa; fuerte asociación con cáncer; mal pronóstico |
| Anticuerpos específicos en miopatía necrosante inmunomediada | |||
| Anti-SRP | Partícula de reconocimiento de señal | 5-15% pacientes MII13-54% pacientes con MNI | Severa debilidad muscular; disfagia; posible compromiso cardiaco; refractaria a inmunosupresores59 |
| Anti-HMGCR | 3-hidroxi-3-metilglutaril-coenzima A reductasa | ∼60% pacientes MNI59 | Miositis con CK muy elevada;con o sin exposición a estatinas; buena respuesta a inmunosupresores59 |
| Anticuerpos específicos en miopatía por cuerpos de inclusión | |||
| Anti-cN1A | 5’nucleotidasa 1A citosólica | 30-60% MCI | Miositis con debilidad asimétrica, compromiso bulbar, respiratorio y mayor mortalidadPresentes también en LES, síndrome de Sjögren, otras |
AEM: anticuerpos específicos de miositis; CK: creatina quinasa; DM: dermatomiositis; DMCA: dermatomiositis clínicamente amiopática; DMJ: dermatomiositis juvenil; EPI: enfermedad pulmonar intersticial; EPI-RP: enfermedad pulmonar intersticial rápidamente progresiva; LES: lupus eritematoso sistémico; MCI: miopatía por cuerpos de inclusión; MII: miopatía inflamatoria inmunomediada; MNI: miopatía necrosante inmunomediada.

Los AEM clásicos son los anti-aminoacil-ARNt sintetasa (AAS), anti-Mi-2 (complejo de desacetilasa remodelador de nucleosomas) y anti-SRP (partícula de reconocimiento de señal). En la última década se identificaron nuevos AEM como el anti-TIF1-γ (factor intermediario de transcripción 1 γ), anti-NXP-2 (proteína de matriz nuclear 2), anti-MDA5 (gen 5 asociado a la diferenciación del melanoma), anti-SAE (enzima activadora de SUMO) y anti-HMGCR (3-hidroxi-3-metilglutaril-CoA reductasa)7–9. Los anticuerpos Anti-cN-1A (5’nucleotidasa 1A citosólica) fueron los últimos que demostraron utilidad clínica para la estratificación de pacientes11.
Estos anticuerpos raramente se encuentran en otras enfermedades autoinmunes y tienen una fuerte asociación con fenotipos clínicos distintivos12.
Anticuerpos específicos en el síndrome anti-sintetasaLos anticuerpos presentes en el SAS pertenecen a la clase IgG13 y su antígeno diana son un grupo de enzimas citoplasmáticas que catalizan la unión de cada aminoácido a su ARNt específico durante la síntesis proteica, llamadas aminoacil-ARNt-sintetasas (AAS). Se unen con los epítopes estructurales de las enzimas para inhibir su actividad11, pero su patogenicidad no ha sido aún demostrada14. Estos anticuerpos son conocidos como anticuerpos anti-AAS o anti-sintetasa. Hasta ahora se han identificado 8 anticuerpos anti-sintetasa: anti-histidil-ARNt sintetasa (anti-Jo-1), anti-treonil-ARNt sintetasa (anti-PL-7), anti-alanil-ARNt sintetasa (anti-PL-12), anti-glicil-ANRt sintetasa (anti-EJ), anti-isoleucil-ARNt sintetasa (anti-OJ), anti-asparaginil-ARNt sintetasa (anti-KS), anti-fenilalanil-ARNt sintetasa (anti-ZO) y anti-tirosil-ARNt sintetasa (anti-YRS/HA)15. El primer anticuerpo descrito de este grupo fue el Jo-1 en 1980 y es el más frecuente de ellos11.
El SAS es una entidad clínico/serológica definida por la presencia de uno de los anticuerpos anti-sintetasa asociado con las siguientes características clínicas que pueden combinarse de diversas formas a lo largo del curso de la enfermedad: miositis, enfermedad pulmonar intersticial (EPI), manos de mecánico, artritis, fenómeno de Raynaud, y fiebre. Los anticuerpos anti-sintetasa se pueden encontrar con una variabilidad del 11 al 39,19% de los pacientes con MII según las series16. Los anti-Jo-1 se detectan en aproximadamente del 20 al 30% de los pacientes con miositis, es el autoanticuerpo más frecuente y ampliamente disponible en la mayoría de los ensayos comerciales, mientras que el resto de los anticuerpos anti-sintetasa son menos comunes y cada uno alcanza menos del 5% de prevalencia en MII8,16.
La detección de estos anticuerpos tiene implicancias diagnósticas, pronósticas y terapéuticas13,16. La afectación muscular es más severa en pacientes anti-Jo-1 y la EPI es más frecuente en pacientes con anticuerpos anti-PL-12 y anti-PL-717.
Los anticuerpos anti-Ro (incluido Ro52) se consideran el tipo más común de AAM en pacientes con SAS y se encuentran con una variabilidad del 30 al 65% de los casos16.
Anticuerpos específicos en dermatomiositisLos anticuerpos específicos en dermatomiositis (AEDM) son potencialmente útiles por múltiples motivos: pueden facilitar el diagnóstico en ausencia de una biopsia muscular patológica en pacientes con DM atípica y aunque se desconoce si son efectivamente patogénicos, tienen valor pronóstico, pueden guiar el tratamiento y eventualmente pueden servir en la selección de pacientes para estudios clínicos basados en serología.
Hasta la fecha, se han identificado 5 AEDM: anti-Mi-2, anti-MDA5, anti-TIF1, anti-NXP-2 y anti-SAE15.
A continuación, se describen la historia, las características y la utilidad clínica de cada uno de estos anticuerpos individualmente:
Anticuerpos anti-Mi-2Fueron descritos por primera vez en una paciente con DM en el año 197618.
Pertenecen a la clase IgG cuyo antígeno diana, la proteína Mi-2, es una helicasa nuclear, que forma parte del complejo núcleo-desacetilasa de remodelación (NuRD) implicado en la transcripción génica.
La proteína Mi-2 está sobreexpresada durante la regeneración muscular en pacientes con DM, lo que sugiere un papel del anticuerpo anti-Mi-2 en la patogenia de la enfermedad. Por otro lado, se demostró que Mi-2 es esencial en la reparación de la epidermis basal, por lo que estaría relacionada con la afectación cutánea, además se vio que se encuentra sobre expresada en los queratinocitos humanos tras la exposición a la luz ultravioleta (UV), por lo que se piensa que la luz UV podría estar relacionada con el inicio de la DM. Otro factor asociado a los anticuerpos anti-Mi-2 es su fuerte asociación con HLA (DRB1*0302 y DRB1*0701)8,19.
Estos anticuerpos se detectan comúnmente en los pacientes con DM del adulto y juvenil con una frecuencia del 11 al 59% y del 4 al 10% respectivamente, según las series8,14.
Clínicamente los pacientes con anticuerpos anti-Mi-2 presentan manifestaciones principalmente cutáneas que incluyen pápulas de Gottron, eritema heliotropo, erupciones con el signo de V, el signo del chal e hipertrofia de cutículas. El pronóstico es más favorable debido a que el compromiso muscular es leve, tienen menor riesgo de EPI y baja frecuencia de asociación con cáncer, así como una adecuada respuesta a la terapia inmunosupresora8,10,11,19.
Anticuerpos anti-SAEFueron descriptos en el año 2007 en pacientes con DM20. Su antígeno diana es la enzima activadora de SUMO (pequeño modificador similar a la ubiquitina), un heterodímero compuesto por 2 subunidades, SAE-1 y SAE-2, involucrado en la modificación post transcripcional de proteínas8.
Los anticuerpos anti-SAE están asociados a un fenotipo típico de DM, se encuentran presentes en aproximadamente el 8% de los adultos, aunque la prevalencia varía según se trate de cohortes de pacientes europeos (4-10%) o asiáticos (del 1-3%), posiblemente vinculado a su asociación genética con los haplotipos HLA DRB1*04-DQA1*03-DQB1*038,20,21. Los pacientes que presentan este anticuerpo manifiestan compromiso cutáneo severo, afectación miopática leve al inicio, pero desarrollan miopatía y disfagia severa posteriormente. De todas maneras, los anticuerpos anti-SAE se asocian a pronóstico favorable debido a que el compromiso pulmonar y la asociación con cáncer son raros8,12,20,21.
Anticuerpos anti-MDA-5Fueron descritos en el año 2005 por Sato et al. en los pacientes con DM amiopática22. Su antígeno diana, la proteína MDA5, es una helicasa específica de ARN involucrada en la respuesta inmune antiviral21.
Los autoanticuerpos contra MDA5 se identifican en la mayoría de adultos y niños con DM clínicamente amiopática (DMCA) y en el 10 al 30% de los pacientes con DM en general21–23.
Clínicamente, los pacientes con DM anti-MDA-5 positivos presentan inflamación muscular de bajo grado o ausente, afectación cutánea grave como úlceras cutáneas, pápulas palmares (pápulas de Gottron inversas), alopecia, paniculitis25 y, además, EPI rápidamente progresiva (EPI-RP) aguda o subaguda. Hasta ahora no hay evidencia de asociación con cáncer21,24. Los niveles elevados de ferritina sérica podrían estar asociados con un mal pronóstico de EPI-RP en pacientes con DM anti-MDA526.
Anticuerpos anti-TIF1-γTIF1 es una proteína supresora de tumores que se encarga de actuar como co-represor transcripcional. Contiene tres subunidades (α, β y γ), cada una tiene sus propios autoanticuerpos. Estos anticuerpos fueron identificados por primera vez en el año 2006 por Targoff y simultáneamente por Kaji, y se encuentran específicamente en la DM (del 15 al 20% de los casos de DM en adultos y en el 20% de los casos de DMJ)27.
Dos tercios de los pacientes con DM anti-TIF-1 presentan autoanticuerpos anti-TIF-1γ y anti-TIF-1α, mientras que el tercio restante es positivo para autoanticuerpos anti-TIF-1γ, exclusivamente. Aunque se afirma que los AEM son mutuamente excluyentes, se han descrito pacientes doblemente positivos para anticuerpos anti-TIF-1α/Mi-28. Clínicamente, los pacientes anti-TIF-1 positivos pueden clasificarse en 2 grupos de acuerdo a la edad: 1) pacientes menores de 40 años, con una DM clásica y 2) pacientes mayores de 40 años, con miositis asociada a cáncer, siendo los tumores sólidos, como el cáncer de ovario, pulmón y mama las neoplasias más comúnmente asociadas, aunque también se han descriptos trastornos hematológicos8.
En general, los pacientes anti-TIF-1γ presentan una DM hipomiopática con baja prevalencia de compromiso sistémico, como EPI, fenómeno de Raynaud y artritis. La afectación cutánea generalizada presenta características únicas, como pápulas hiperqueratósicas palmares, dermatitis de tipo psoriásico, placas atróficas hipopigmentadas con telangiectasias y parches palatinos ovoides8.
El riesgo de malignidad es mayor en pacientes con anti-TIF-1γ/α que en aquellos con anti-TIF-1γ solo8,19. Esta asociación no se confirmó en niños o pacientes adultos jóvenes11.
Anticuerpos anti-NXP-2NXP-2 es una proteína involucrada en múltiples funciones nucleares, incluida la regulación de la transcripción y el metabolismo del ARN. Los anticuerpos anti-NXP-2 (antes anti-MJ) se asociaron inicialmente con DMJ grave, complicada por calcinosis, poliartritis y vasculitis intestinal21,28. Recientemente, también fueron encontrados en pacientes adultos, con una prevalencia variable del 1,6 al 17%. Esta variabilidad en la prevalencia puede atribuirse a diferencias étnicas y/o en los métodos de detección de anticuerpos utilizados8,21,28. Estudios recientes asociaron estos anticuerpos con elevado riesgo de malignidad subyacente y debilidad distal atípica para DM21,28.
Los anticuerpos anti-NXP2 son el segundo autoanticuerpo más común en pacientes con DMJ con una prevalencia del 20 al 25%21,28. La DMJ con anticuerpos anti-NXP2 se asocia a miopatía severa, consecuencia de la isquemia muscular inducida por vasculopatía21, que lleva al desarrollo de contracturas, tiene un mal pronóstico y requiere un manejo más agresivo que otras formas de DMJ21.
Anticuerpos específicos en miopatía necrosante inmunomediadaDebido a que con frecuencia la MNI se diagnostica erróneamente como polimiositis, es difícil establecer la proporción de pacientes con MII que tiene MNI, pero se estima que representa del 17 al 45% del total de las MII29. Puede presentarse a cualquier edad, pero se observa principalmente en adultos30.
En la actualidad, según la positividad de los anticuerpos específicos, la MNI se categoriza en tres subgrupos: SRP, HMGCR y seronegativa17. Existen evidencias que apuntan a un rol potencialmente patogénico de los mismos en el desarrollo de la MNI29. Los niveles de los anticuerpos tendrían relación con la gravedad de la enfermedad y con la actividad de la creatinfosfoquinasa (CPK)31,32.
Esta enfermedad se caracteriza por debilidad muscular proximal y marcada elevación de la CPK. Suele ser más severa en pacientes jóvenes y puede progresar crónicamente simulando una distrofia muscular17.
La MNI seronegativa, comparada con las seropositivas, se asocia más frecuentemente a enfermedades del tejido conectivo, enfermedades extramusculares y mayor riesgo de cáncer17–34.
Múltiples publicaciones apoyan el uso de autoanticuerpos anti-SRP y anti-HMGCR en el diagnóstico y clasificación de las MNI33.
Anticuerpos anti-SRPEn 1986 se describen por primera vez anticuerpos anti-SRP en el suero de un paciente con «polimiositis»35.
La SRP es una ribonucleoproteína citoplasmática formada por 6 cadenas polipeptídicas unidas a una pequeña molécula de ARN, y es esencial para la translocación de polipéptidos nacientes en el retículo endoplásmico36.
La prevalencia de autoanticuerpos anti-SRP varía del 5 al 15% en pacientes con MII y del 13 al 54% en pacientes con miopatía nnecrosante27,33.
El 70% de los pacientes anti-SRP presentan severa debilidad muscular, de rápida progresión, de distribución típicamente simétrica, afectando específicamente músculos proximales y axiales. El 7% muestra un síndrome de cabeza caída. La disfagia también es un síntoma frecuente (41% de los casos)33,36, al igual que el compromiso cardiaco37. La EPI se presenta en el 13% de los casos. Es rara la existencia de otros síntomas sistémicos36,37.
Anticuerpos anti-HMGCRLa HMGCR es una enzima intracelular localizada en el retículo endoplásmico que controla la síntesis de colesterol. Es la misma enzima bloqueada farmacológicamente por las estatinas.
Inicialmente se describieron autoanticuerpos contra una banda compleja de 200/100 kDa en pacientes con MNI, y solo después se identificaron como autoanticuerpos anti-HMGCR38.
No es obligatorio un antecedente de exposición a estatinas para desarrollar MNI anti-HMGCR39. Este antecedente se encontró en el 38 al 63% de los pacientes con MNI anti-HMGCR positivos, principalmente en pacientes mayores. Estos anticuerpos también se vieron asociados con un mayor riesgo de cáncer40,41.
Los títulos de anticuerpos anti-HMGCR se asociaron con la actividad de la CK y se relacionaron inversamente con la fuerza muscular42.
Los pacientes anti-HMGCR positivos presentan una MNI típica, responden bien a la terapia inmunosupresora y a las inmunoglobulinas intravenosas, pero tienden a recaer después de la reducción gradual. Más del 50% de aquellos que recuperaron la fuerza total continuaron teniendo niveles de CK superiores a 500 UI/l39.
Los pacientes más jóvenes experimentan una enfermedad más grave con peor pronóstico39,42.
Anticuerpos específicos en miopatía por cuerpos de inclusión esporádica (MCIe)La MCIe, a diferencia de otras miopatías inflamatorias, es más frecuente en hombres, afecta a pacientes mayores de 50 años y su evolución es lentamente progresiva, con un patrón de debilidad característicamente asimétrico, predominantemente en cuádriceps, flexores profundos de los dedos y flexores de muñeca30. Estos pacientes también pueden presentar disfagia progresiva30.
La MCIe no se asocia con ningún autoanticuerpo específico de miositis, pero en el 30 al 60% de los casos puede detectarse la presencia de anticuerpos anti-cN1A. Estos anticuerpos también se pueden encontrar en el 5 al 10% de los pacientes con polimiositis, en el 20% de los pacientes con DM, en el 10% de los pacientes con lupus eritematoso sistémico y en el 12% con síndrome de Sjögren43,44.
La biopsia muscular de estos pacientes a menudo incluye inflamación, disfunción mitocondrial, agregación anormal de proteínas y presencia de vacuolas lineadas. Aunque estas vacuolas también pueden encontrarse en otras miopatías hereditarias, su presencia puede ayudar a distinguir la MCIe de otras miopatías inflamatorias.
Por microscopía electrónica pueden evidenciarse inclusiones tubo-filamentosas que dieron lugar al nombre de miositis por cuerpos de inclusión.
No existe evidencia clara que muestre que la inmunosupresión beneficie a los pacientes con MCIe6,30.
Anticuerpos anti-cN1AFueron descriptos por primera vez en el 2011 por Salajegheh et al.42. El autoantígeno es la enzima muscular 5’nucleotidasa 1A citosólica (cN-1A) que es una proteína implicada en el metabolismo de los ácidos nucleicos45,46. Se desconoce el papel de los anticuerpos anti-cN1A en la patogénesis de MCIe46.
Estos anticuerpos tienen una sensibilidad del 33 al 76% y una especificidad del 92 al 96% para MCIe45–47. A pesar de la alta especificidad observada inicialmente, estos anticuerpos fueron luego detectados en pacientes con otros trastornos autoinmunes: en el síndrome de Sjögren (23 al 36%), en el lupus eritematoso sistémico (14 al 20%) y en DM (15%)43,44,46. Por tanto, su presencia debe interpretarse con cautela, teniendo en cuenta el contexto clínico y los hallazgos histopatológicos del paciente.
La positividad de este anticuerpo en pacientes con MCIe, se asoció a mayor debilidad muscular, compromiso bulbar, respiratorio y mayor mortalidad17,48.
Autoanticuerpos asociados a miositis (tabla 3)Los AAM incluyen anticuerpos anti-Ro/SSA, anti-PM/Scl, anti-Ku y anti-U1-RNP.
Asociaciones clínicas y frecuencia de AAM
| Anticuerpo | Autoantígeno | Frecuencia | Presentación clínica |
|---|---|---|---|
| Anti-Ro/SSA | Proteínas 52 y 60kDa asociadas a ARN | >30% en pacientes con MII | Asociado a anti-Jo1: miositis severa con compromiso articular; mayor riesgo de EPI y cáncer |
| Anti-PM/Scl | Complejo nucleolar macromolecular involucrado en la degradación del ARNm | 4-12% MII adultos5% MII jóvenes | Sme superposición PM/DM y esclerosis sistémica; fenómeno de Raynaud, artritis, manos de mecánico, disfagia y EPI |
| Anti-Ku | Heterodímero de 2 subunidades de 70 y 80KDa | 1,3-50% pacientes con MII | Miositis, fenómeno de Raynaud, artralgia, EPI |
| Anti-U1RNP | Pequeña ribonucleoproteína nuclear llamada U1 snRNP | 3-8% MII25-40% en smes de superposición | Debilidad proximal con preservación del cuello, fenómeno de Raynaud, artritis, rash, disfagia, EPI y pericarditis |
AAM: anticuerpos asociados a miositis; ARN: ácido ribonucleico; ARNm: ARN mensajero; DM: dermatomiositis; DMJ: dermatomiositis juvenil; EPI: enfermedad pulmonar intersticial; EPI-RP: enfermedad pulmonar intersticial rápidamente progresiva; MCI: miopatía por cuerpos de inclusión; MII: miopatía inflamatoria inmunomediada; MNI: miopatía necrosante inmunomediada; PM: polimiositis; snRNP: ribonucleoproteínas nucleares pequeña.
Se encuentran a menudo cuando la miositis ocurren como un componente de otra enfermedad del tejido conectivo (ETC).
Estos anticuerpos están dirigidos contras antígenos expresados en el núcleo (anti-Ku, anti-PM/Scl) o el citoplasma de la célula (anti-Ro/SSA).
Anticuerpos anti-Ro/SSALos anticuerpos anti-Ro/SSA son inmunoglobulinas que se dirigen contra proteínas 52 kDa y 60 kDa asociadas a ARN. Ro52 es bioquímica e inmunológicamente distinto de Ro60 y tiene la mayor inmunogenicidad49.
Los anticuerpos anti-Ro52 no son específicos para ninguna enfermedad, ya que pueden estar presentes en ETC o en otras condiciones, como infecciones virales, enfermedades neoplásicas e incluso en individuos sanos49. A pesar de su baja especificidad, son los autoanticuerpos más frecuentes del grupo AAM, y están presentes en más del 30% de los pacientes con MII50.
Frecuentemente se asocian a AEM (Ac anti-Jo-1, anti-MDA5, anti-SRP). La asociación con el anticuerpo anti-Jo-1 está relacionada con un fenotipo de miositis de curso severo y compromiso articular49,51.
Además, la presencia de anticuerpos anti-Ro se asocia a mayor riesgo de EPI, compromiso articular y cáncer49,51.
Anticuerpos anti-PM/SclEstos autoanticuerpos se dirigen contra un complejo nucleolar macromolecular compuesto por varias proteínas cuya principal función es la degradación del ARNm. De este complejo, las 2 principales proteínas autoantigénicas son la PM/Scl-75 y la PM/Scl-10052.
Estos anticuerpos están presentes en aproximadamente el 4 al 12% de los pacientes adultos y en el 5% de los jóvenes con MII7,8.
Los anticuerpos anti-PM/Scl pueden considerarse de alguna manera marcadores del síndrome de superposición entre PM/DM y esclerosis sistémica, y marcarían un aumento en el riesgo de presentar fenómeno de Raynaud, artritis, manos de mecánico, disfagia y EPI8,10,53.
En cuanto al compromiso muscular, se observó un patrón de distribución proximal, más severo en miembros superiores, con marcada atrofia de deltoides.
Anticuerpos anti-KuFueron descritos por Mimori et al. en 1981. El antígeno fue denominado Ku por las iniciales del paciente de quien se utilizó el suero para identificarlo54. Este antígeno es un heterodímero de 2 subunidades de 70 y 80 KDa que se une a los extremos libres del ADN y juega un papel clave en la reparación del mismo55.
Se describen miopatías inflamatorias con anticuerpos anti-Ku, principalmente en el contexto de un síndrome de superposición55. La prevalencia de anticuerpos anti-Ku reportados en la literatura varían considerablemente oscilando entre el 1,3 y el 50%55. En series de pacientes anti-Ku positivos se informan características clínicas como fenómeno de Raynaud, artralgia y miositis55,56. Los pacientes anti-Ku con miositis frecuentemente asocian EPI56.
Anticuerpos anti-U1-snRNPEstos anticuerpos se dirigen contra una pequeña ribonucleoproteína nuclear llamada U1-snRNP. Esta ribonucleoproteína es uno de los complejos ARN-proteína que participa en el procesamiento del pre-ARNm en el núcleo, removiendo la mayoría de los intrones57.
Los pacientes con anticuerpos anti-U1-RNP presentan debilidad muscular proximal con preservación de los músculos del cuello y aproximadamente el 57% de sus biopsias musculares muestran características necrosantes58.
Dentro de las manifestaciones extramusculares, se incluyen fenómeno de Raynaud (80%), artralgia/artritis (60%), características cutáneas de DM (60%), manos de mecánico (50%) y disfagia (50%). Otras manifestaciones clínicas frecuentes en estos pacientes son EPI (45%), pericarditis (40%), edema subcutáneo (35%), fiebre (35%), glomerulonefritis (25%), hipertensión pulmonar (25%), esclerodactilia (25%) y calcinosis (25%)58.
Una alta proporción de pacientes anti-U1-RNP-positivos son afroamericanos57.
Su prevalencia es baja (3-8%) en los pacientes con miositis61, pero aumenta en los síndromes de superposición (25-40%), principalmente EMTC, lupus eritematoso sistémico y esclerosis sistémica. Generalmente suele tener buen pronóstico19,59.
ConclusiónEl diagnóstico de las MII es a menudo un desafío, debido a su heterogeneidad. La posibilidad de identificar AEM y AAM contribuirá a facilitar el diagnóstico, conocer el pronóstico y de esta manera planificar más adecuadamente el tratamiento de las MII.
Nuevas clasificaciones de MII con la inclusión de criterios serológicos, permitirán definir poblaciones de pacientes más homogéneas y mejorará el poder de los ensayos clínicos para identificar los tratamientos dirigidos a los diferentes subgrupos de pacientes.
A medida que se amplía el conocimiento de los mecanismos patogénicos involucrados en estas enfermedades, esperamos que se identifiquen dianas terapéuticas adicionales.
Responsabilidades éticasSe declara, el haber respetado los principios éticos de investigación.
FinanciaciónLos autores declaran no haber recibido financiación para la realización de este trabajo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses
