




La paraparesia espástica (PPE) crónica es un motivo común de consulta neurológica. Algunos hallazgos al examen físico pueden orientar a etiologías en particular.
ObjetivoEl objetivo de este trabajo fue describir las características clínicas de una serie de pacientes con PPE crónica.
Pacientes y métodosSe realizó un estudio retrospectivo observacional en 46 pacientes con PPE crónica, evaluados entre 2005 y 2019 en el departamento de Neurología de nuestro hospital. Se consignaron variables epidemiológicas, clínicas y los estudios diagnósticos realizados.
ResultadosHubo 46 pacientes con dicha sintomatología. La media de edad fue de 43,64años (±18,02) y el 65,2% fueron hombres. La mayoría (80,4%) presentaron hiperreflexia de miembros superiores. Los trastornos esfinterianos (56,22%) y el déficit sensitivo (47,8%) también fueron frecuentes. Solo se obtuvo diagnóstico etiológico en un tercio de los pacientes.
ConclusionesLa PPE es frecuente que tenga síntomas asociados. Los principales síntomas acompañantes fueron en este estudio la hiperreflexia de miembros superiores y los déficits esfinterianos y sensitivos. El diagnóstico definitivo se logró en un tercio de la población, menor a otros estudios publicados.
Spastic paraparesis (SPP) is a common cause for neurological consultation. Some signs or symptoms are more frequent than others and may point to a specific etiology.
ObjectiveThe aim of this study is to describe the clinical characteristics of a group of patients with chronic SPP.
Patients and methodsA retrospective observational study was carried out in 46 patients with SPP, who were evaluated between 2005 and 2019. We studied epidemiological, diagnostic and clinical variables.
ResultsThere were 46 patients with SPP, 63.04% of which were male with a mean age of the patients was 43.64 (±18.02) years. Most patients (80.4%) had upper limb hyperreflexia besides SPP. Sphincter disorders (56.22%) and sensory abnormalities (47.8%) were also frequent. Diagnosis was obtained in only one third of our patients.
ConclusionsIsolated SPP is an infrequent finding. The most common concomitant signs or symptoms are upper limb hyperreflexia, sphincter and sensory abnormalities. In our sample, a definite diagnosis was obtained in only a third of the cases, lower than other published series.
La paraparesia espástica (PPE) crónica es una causa común de consulta neurológica. Las etiologías descriptas son numerosas y son necesarios muchos estudios complementarios para arribar al diagnóstico. Sus causas son adquiridas o genéticas (con formas de herencia autosómicas dominantes, autosómicas recesivas, ligadas alX, mitocondriales o esporádicas, estas últimas representando del 13 al 40% de los casos genéticos)1. Dentro de las formas adquiridas se incluyen las infecciosas (por HTLV-1, HIV o sífilis), las metabólicas, las inflamatorias (esclerosis múltiple, espectro neuromielitis óptica), las estructurales (compresivas), las degenerativas (enfermedades de motoneurona) y las vasculares2-4.
Las PPE hereditaria es un grupo heterogéneo de entidades que posee una prevalencia mundial combinada de 2-5 casos cada 100.000 personas5. La clasificación genética se basa en la secuenciación de loci cromosómicos de genes específicos, conocidos como SPG (del inglés spastic paraplegia gene). Generalmente se las clasifica en formas «puras» y «complicadas»1,6. Ambas comparten espasticidad simétrica asociada a hiperreflexia en miembros inferiores (MMII), con reflejos plantares extensores. Generalmente dentro de las formas «puras» se incluye sintomatología no motora: hipopalestesia (generalmente presente desde el inicio de la enfermedad) y trastornos esfinterianos (mayoritariamente de presentación más tardía)1,6,7. Otros síntomas neurológicos, como deterioro cognitivo, ataxia, disartria, neuropatía y crisis comiciales, se encuentran reportados en el 50% de los casos y suelen incluirse dentro de las formas complicadas1. Algunas comorbilidades permiten esbozar una aproximación diagnóstica hacia los posibles genes alterados. La masivización de nuevas formas de estudio genético (NGS [de las siglas en inglés next generation sequencing]) que permiten la detección de anomalías en paneles de genes o incluso en el genoma completo probablemente permita mejorar la proporción de pacientes con o sin antecedentes familiares que hoy en día son diagnosticados (51-71%)8-10. La ataxia espinocerebelosa tipo3 (SCA3-MJD) siempre debe ser considerada como diagnóstico diferencial, dado que puede progresar con PPE y es una entidad frecuente de etiología genética11.
Las causas metabólicas incluyen el déficit de vitaminaB12, cobre o vitaminaE2,4. Aunque la presentación clínica es similar, los síntomas cerebelosos y la concomitancia de neuropatía periférica generalmente orientan a déficit de vitaminaE y B12, respectivamente.
Dentro de las causas infecciosas de PPE, el virus HTLV-1 es endémico en el noroeste de la Argentina y se encuentra asociado geográfica y étnicamente con las comunidades nativas de esa área12. A nivel mundial existen otras regiones endémicas, principalmente en Sudamérica, el Caribe y Japón. Se calcula que alrededor de 5-10 millones de personas se encuentran infectadas a nivel mundial por este virus, aunque probablemente sea una subestimación, dada la falta de estudios epidemiológicos adecuados13,14. Sin embargo, solo un 2% de ellos presentarán complicaciones neurológicas15. El cuadro clínico de estos pacientes se caracteriza por compromiso esfinteriano y dolor lumbar16, siendo la presencia de nivel sensitivo un hallazgo excepcional15,17. No cursa con signos o síntomas patognomónicos, por lo que la sospecha diagnóstica basada en la epidemiología debe orientar a su búsqueda serológica en plasma y líquido cefalorraquídeo (LCR). La segunda etiología infecciosa viral corresponde a la asociada al HIV, que puede ocasionar una mielopatía vacuolar. Los síntomas son de aparición tardía en la infección por HIV18, con una cronología que comienza con trastornos esfinterianos, adiciona PPE y finalmente déficit sensitivo. Es poco frecuente el compromiso de la sensibilidad termoalgésica o la presencia de dolor, y puede cursar con neuropatía periférica, como ocurre en el déficit de vitaminaB1219,20.
Hasta en el 75% de los casos de HIV puede haber coinfección con Treponema pallidum y los pacientes pueden desarrollar neurosífilis aun si completaron tratamiento para sífilis primaria21,22. Por lo tanto, la neurosífilis siempre debe considerarse como diagnóstico diferencial en pacientes HIV con sintomatología neurológica. Más aun, la meningomielitis sifilítica puede manifestarse como PPE de lenta evolución asociada a trastornos esfinterianos y sensitivos (hipopalestesia e hipocinestesia). Típicamente estos pacientes presentan afección de otras topografías del sistema nervioso central, por lo que pueden tener sintomatología florida: ataxia, oftalmoplejía, paresias, etc.22.
Las enfermedades desmielinizantes, como la esclerosis múltiple y aquellas del espectro de la neuromielitis óptica, suelen cursar con lesiones en el tronco encefálico y la médula espinal que pueden determinar diversos síntomas además de la PPE (debilidad, fatiga, síndromes de pares craneales, hipoestesia, etc.)23. Sin embargo, la espasticidad en los MMII (y también en los miembros superiores [MMSS]) es uno de los síntomas más prevalentes (presente hasta en el 80% de los pacientes) y discapacitantes24.
Dentro de las patologías estructurales que pueden presentarse como PPE se encuentran de forma más frecuente la mielopatía cervical espondilótica, los tumores del foramen magno, la hidrocefalia, la siringomielia y los meningiomas parasagitales3,4. Las principales causas degenerativas son la esclerosis lateral amiotrófica y la esclerosis lateral primaria. Estas entidades pueden presentarse como PPE sin compromiso sensitivo o vegetativo3.
Finalmente, también etiologías vasculares como la trombosis de la arteria cerebral anterior, las trombosis venosas cerebrales y las malformaciones arteriovenosas medulares pueden presentarse como PPE con síntomas progresivos3,4.
El objetivo de este estudio es describir las características clínicas en una serie de 46 pacientes con PPE crónica sin antecedente traumático.
Materiales y métodosSe desarrolló un estudio retrospectivo observacional en el departamento de Neurología del Hospital Ramos Mejía de la ciudad de Buenos Aires, Argentina. Fueron incluidos todos aquellos pacientes evaluados en el servicio entre los años 2005 y 2019 por PPE crónica no traumática (definida como hipertonía y debilidad en los MMII de inicio insidioso). Se registraron variables epidemiológicas (sexo, edad, nacionalidad, tiempo desde el inicio de los síntomas a la primer consulta hospitalaria), diagnósticas (serología para HIV y HTLV-1, VDRL, resonancia nuclear magnética [RNM] de cerebro y columna, análisis de LCR, electromiograma [EMG] y test genéticos) y clínicas (tono, fuerza muscular, reflejos, clonus, taxia, marcha, trastornos esfinterianos, déficit cognitivo, neuropatía craneal). La fuerza muscular fue objetivada según la escala del Medical Research Council. Se consideró deterioro cognitivo cuando el paciente puntuó menos de 24 puntos en el Minimental Test de Folstein. Se consideró patología esfinteriana cuando el paciente refirió incontinencia urinaria o fecal, urgencia miccional o constipación.
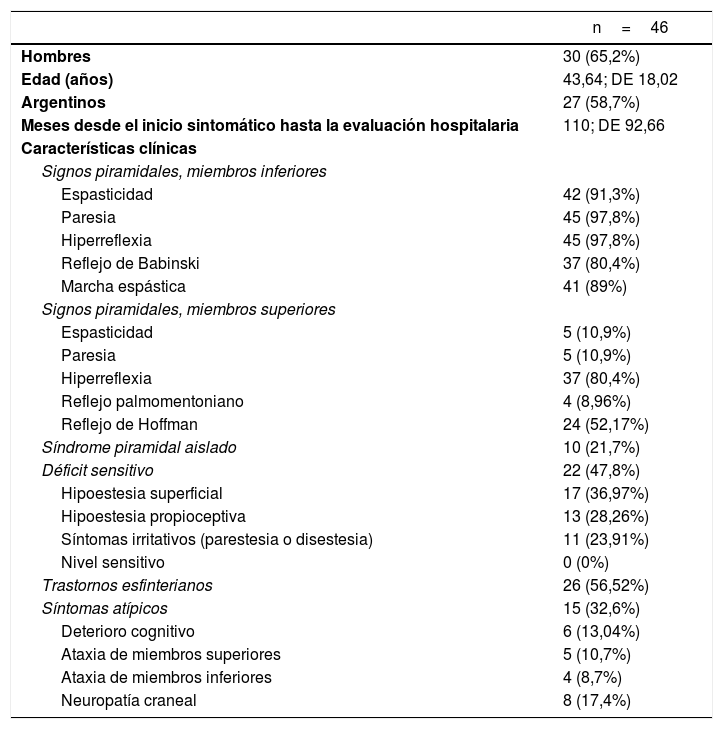
ResultadosSe incluyeron 46 pacientes con PPE. La mayoría fueron de sexo masculino (n=30; 65,2%) y argentinos (n=27; 58,7%). La edad media fue de 43,64años (DE±18,02) y la media de tiempo entre el inicio de síntomas y la consulta hospitalaria fue de 110 meses (DE±92,66).
Con relación a las características clínicas en los MMII, al menos dos signos piramidales (espasticidad, debilidad, hiperreflexia o reflejos patológicos) se encontraron en todos los pacientes. En todos los pacientes deambuladores (n=41; 89%) se objetivó marcha espástica. Los signos piramidales en los MMSS fueron más heterogéneos: 37 pacientes (80,4%) presentaron hiperreflexia y solo 5 (10,9%) espasticidad o debilidad (2 pacientes presentaban ambos). El reflejo de Hoffman fue positivo en 24 pacientes (52,17%), mayoritariamente como un hallazgo aislado. El hallazgo de un reflejo palmomentoniano fue raro (n=4; 8,96%).
Solo 10 pacientes de la muestra (21,7%) tuvieron como único hallazgo patológico el síndrome piramidal. La mayoría de los pacientes (n=26; 56,52%) refirieron algún grado de trastorno esfinteriano. Con relación a la sensibilidad, 17 pacientes (36,97%) presentaron hipoestesia superficial, 11 (23,91%) síntomas irritativos (parestesias, disestesias) y 13 (28,26%) hipopalestesia. Ninguno presentó nivel sensitivo. En 6 pacientes (13,04%) se objetivó deterioro cognitivo, 5 (10,7%) presentaron ataxia de MMSS y 4 (8,7%) ataxia de MMII. En 8 pacientes (17,4%) se encontró patología concomitante en algún par craneal. Estos hallazgos epidemiológicos y clínicos se encuentran resumidos en la tabla 1.
Características clínicas y demográficas en los pacientes con paraparesia espástica
| n=46 | |
|---|---|
| Hombres | 30 (65,2%) |
| Edad (años) | 43,64; DE 18,02 |
| Argentinos | 27 (58,7%) |
| Meses desde el inicio sintomático hasta la evaluación hospitalaria | 110; DE 92,66 |
| Características clínicas | |
| Signos piramidales, miembros inferiores | |
| Espasticidad | 42 (91,3%) |
| Paresia | 45 (97,8%) |
| Hiperreflexia | 45 (97,8%) |
| Reflejo de Babinski | 37 (80,4%) |
| Marcha espástica | 41 (89%) |
| Signos piramidales, miembros superiores | |
| Espasticidad | 5 (10,9%) |
| Paresia | 5 (10,9%) |
| Hiperreflexia | 37 (80,4%) |
| Reflejo palmomentoniano | 4 (8,96%) |
| Reflejo de Hoffman | 24 (52,17%) |
| Síndrome piramidal aislado | 10 (21,7%) |
| Déficit sensitivo | 22 (47,8%) |
| Hipoestesia superficial | 17 (36,97%) |
| Hipoestesia propioceptiva | 13 (28,26%) |
| Síntomas irritativos (parestesia o disestesia) | 11 (23,91%) |
| Nivel sensitivo | 0 (0%) |
| Trastornos esfinterianos | 26 (56,52%) |
| Síntomas atípicos | 15 (32,6%) |
| Deterioro cognitivo | 6 (13,04%) |
| Ataxia de miembros superiores | 5 (10,7%) |
| Ataxia de miembros inferiores | 4 (8,7%) |
| Neuropatía craneal | 8 (17,4%) |
DE: desvío estándar.
Pudo obtenerse un diagnóstico definitivo únicamente en 15 pacientes (32,6%). La etiología más común fue la enfermedad desmielinizante (n=8; 53,3%), seguida de la patología infecciosa (n=4; dos HTLV-1, una por HIV y otra por sífilis), mientras que la patología estructural, la enfermedad de motoneurona y la genética (SPG11) fueron diagnosticadas en un paciente cada una.
Con relación a los estudios complementarios, mientras que 16 pacientes tuvieron sospecha clínica de PPE hereditaria (en función de su presentación clínica, edad de presentación, familiares afectados, etc.), los estudios genéticos necesarios pudieron ser realizados únicamente en 4 de ellos (uno resultó positivo para SPG11). Se realizó secuenciación exómica en 2 pacientes: uno en un paciente con diagnóstico final de enfermedad desmielinizante y uno sin diagnóstico definitivo.
Ninguno de los pacientes sin diagnóstico final (n=31) tuvo estudios completos: solo 7 (22,5%) realizaron una punción lumbar (normal), 23 (74,2%) RNM de cerebro, 20 (64,5%) RNM espinal, 10 (32,3%) serología para HIV, 8 (25,8%) serología para HTLV-1, ninguno (0%) VDRL y 12 (38,7%) tenían realizado EMG. Solo 5 pacientes (16,1%) no habían realizado al menos dos de los estudios complementarios habituales.
DiscusiónNuestra población fue predominantemente masculina (65,2%), con una edad media de 43,64 (DE±18,02) años. Estos datos son similares a los hallados en la literatura internacional. En Brasil, un estudio retrospectivo que incluyó 166 pacientes con mielopatía no traumática y cuadro clínico de PPE observaron mayoría de hombres (53%), con una edad media de 48,5 (DE±16,2) años25. Por otro lado, en Inglaterra, en un estudio prospectivo que incluyó 585 pacientes con paraparesia y tetraparesia espástica, el 52,32% fueron hombres26. También en un estudio en Estados Unidos que incluyó una serie de 19 pacientes con el mismo diagnóstico se observó una mayoría de hombres (73,78%) y una edad promedio de 46 (DE±2,9) años27. Finalmente, en un análisis retrospectivo realizado en Camerún que incluyó 224 casos de mielopatías no traumáticas en un lapso de 7años se objetivó nuevamente una mayor proporción de hombres afectados (n=87; 60,5%), con una media de edad de 45,4 (DE±1,6) años28. Dado que estos datos son consistentes en distintas poblaciones, podemos asumir que existe una predominancia en hombres y que la edad más frecuente de diagnóstico es entre los 40 y los 50años.
La media de tiempo desde el inicio sintomático y nuestra consulta hospitalaria (110 meses) es mucho mayor que en las otras series reportadas (49,925 y 59meses27, 12 días28). Creemos que esta demora puede encontrarse relacionada con el hecho de que la mayoría de nuestros pacientes fueron evaluados luego de múltiples consultas en otros centros.
Solo 10 pacientes (21,7%) tuvieron un síndrome piramidal aislado. La mayoría (n=26; 56,52%) presentaron algún grado de compromiso esfinteriano. Otras series también reportaron porcentajes elevados (Lekoubou Looti et al.28, 80,7%; Pinto et al.25, 51,2%). Como en nuestra muestra, en dichas series también fue frecuente la objetivación de trastornos sensitivos (89,8 y 57,8% de los casos, respectivamente). Con relación a los signos atípicos (deterioro cognitivo, ataxia apendicular o neuropatía cranial), los observamos casi en un tercio de los pacientes. Esta sintomatología no fue descripta en la serie de Camerún28 y Brasil25, si bien puede inferirse de la serie inglesa de hallarse en un 37,4% (n=2.019) de los casos26. Nuevamente los datos de las distintas series resultan concordantes, permitiendo establecer que los trastornos esfinterianos y el déficit sensitivo son hallazgos comunes y los síntomas extramedulares no son tan infrecuentes.
La proporción de pacientes con diagnóstico definitivo de nuestro estudio (32,6%) es baja en comparación con las otras series (79,5% en Brasil y 81,4% en Inglaterra)25,26. Consideramos que esto es consecuente a los déficits del sistema de salud público en nuestro país, en el contexto de que la PPE es una enfermedad crónica y lentamente progresiva, lo que podría explicar que los pacientes difieran la realización de estudios complementarios burocráticamente dificultosos, dado que la discapacidad que ocasiona la enfermedad es generalmente leve. Asimismo, se encuentra reportado que en países de bajos y medios ingresos los déficits de infraestructura, la distribución inadecuada de recursos y la falta de educación, entre otros factores, determinan demoras en los diagnósticos y menores oportunidades terapéuticas para los pacientes29-31. Estimamos que esta serie de casos representa dicha realidad.
Una de las limitaciones de este estudio es el sesgo de selección, dado que únicamente incluimos pacientes que consultaron a nuestro departamento de Neurología. Esto podría haber determinado una sobreestimación de los pacientes con síntomas extramedulares, dado que es más común que los mismos sean referidos al neurólogo en lugar de ser abordados por otros especialistas (traumatólogos, neurocirujanos, etc.). Además, la baja frecuencia de diagnóstico definitivo impidió la búsqueda de asociaciones entre la signo-sintomatología y las distintas etiologías. No obstante, consideramos valioso describir nuestra experiencia y nuestras dificultades, dado que la práctica asistencial no siempre es tan lineal como se describe en las publicaciones29-31.
En conclusión, la PPE crónica no traumática en la población estudiada es levemente más frecuente en hombres y suele diagnosticarse entre los 40 y los 50años. Aunque la PPE sea la signo-sintomatología más llamativa, raramente es un hallazgo aislado. Generalmente se asocia a hiperreflexia de MMSS, trastornos esfinterianos y déficit sensitivo. En nuestra serie la realización de estudios complementarios para alcanzar el diagnóstico etiológico fue muy dificultosa, probablemente como consecuencia de las características lentas y progresivas de la propia enfermedad asociadas a problemas estructurales de nuestro sistema de salud.
Consideraciones éticasEste trabajo fue aprobado por el comité de Ética del Hospital Ramos Mejía. En vista de la naturaleza retrospectiva del estudio, los procedimientos y consultas fueron parte del seguimiento habitual de los pacientes.
Conflicto de interesesNo hay conflictos para declarar.
