






El síndrome de Sjogren es una enfermedad autoinmune que se caracteriza por infiltración celular a glándulas salivares, lagrimales, órganos viscerales y vasculares. Las manifestaciones neurológicas pueden llegar a ser poco frecuentes. Por lo que su diagnóstico requiere cumplir determinados criterios. El tratamiento adecuado puede mejorar la clínica del paciente. Se reporta una serie de casos de manifestaciones del sistema nervioso periférico y central de un hospital de referencia nacional.
Sjogren's syndrome is an autoimmune disease characterized by cellular infiltration of the salivary and lacrimal glands, visceral organs, and the vascular system. Neurological manifestations may become rare. Therefore, its diagnosis requires meeting certain criteria. Appropriate treatment can improve the patient's clinic. We present a series of cases of manifestations of the peripheral and central nervous system from a national referral hospital.
El síndrome de Sjogren (SS) es una enfermedad autoinmune que puede ser primaria o secundaria a otra enfermedad del tejido conectivo (lupus eritematoso sistémico, artritis reumatoide o esclerodermia)1. Este síndrome está caracterizado por una infiltración mononuclear y destrucción de las glándulas salivares y lagrimales los cuales conducen a xerostomía y xeroftalmia. La infiltración mononuclear invade órganos viscerales o lesiones vasculíticas que pueden originar manifestaciones extraglandulares2.
Las manifestaciones neurológicas son una de las manifestaciones extraglandulares. Se estima que la presencia de estas manifestaciones se encuentra en el 8,5-70% de los pacientes diagnosticados con SS. En el 25-60% los síntomas neurológicos preceden el diagnóstico de SS durante 2 años en promedio3.
Las manifestaciones neurológicas pueden afectar el sistema nervioso central (SNC) o el sistema nervioso periférico (SNP). La prevalencia de afectación del SNP es del 5-20%, mientras que del SNC es 1-5%4.
Son escasos los reportes de compromiso neurológico del SS en Latinoamérica, más aún en Perú. Previa aprobación del comité de ética de nuestra institución se presentan una serie de casos de manifestaciones neurológicas del SS en un hospital nacional peruano.
Descripción de los casos clínicosCaso 1Paciente mujer de 35 años, con antecedente médico de gastritis crónica. Con una duración de la enfermedad de 6 meses, caracterizado inicialmente por parestesias en manos, agregándose ageusia, disartria, parálisis facial bilateral y al presentar hemiparesia izquierda, fue hospitalizada en otra institución, donde fue diagnosticada como probable síndrome de Miller Fisher, por lo que recibió inmunoglobulina endovenosa durante 4 días, con mejoría parcial. La sintomatología permaneció estacionaria durante un mes, posteriormente se añadió disminución de fuerza muscular en miembros inferiores de forma ascendente, el cual originaba limitación a la deambulación, por lo que empezó a usar silla de ruedas. Ingresa en urgencias de un hospital nacional al persistir parestesias distales y disminución de fuerza en miembros inferiores.
En el examen físico se encontró Glasgow 15/15, despierta, alerta, orientada, concentra, habla fluente, nómina, movimientos oculares conservados, parálisis facial bilateral, fuerza muscular en miembros superiores (MMSS) derecho 4-/4/4, izquierdo 4-/4/4, miembros inferiores (MMII) derecho 4-/4/4, izquierdo 4-/4/4, hiporreflexia MMSS y arreflexia MMII, apalestesia en yemas de dedos, piernas y pies, reflejo cutáneo plantar flexor, lasegue negativo, ataxia sensorial en MMII, sensibilidad conservada, sin signos meníngeos, marcha atáxica.
Durante su evaluación se planteó como primera posibilidad una polineuropatía crónica inflamatoria, por lo que se estableció un plan de trabajo extenso, destacando entre ellos, estudios complementarios como hemograma, perfil bioquímico, electrólitos y proteína C reactiva (PCR) normales, VSG 95, perfil hepático normal, vitamina B12 y ácido fólico normal, perfil tiroideo normal, VHB-VHC negativo, TORCH negativo, HIV negativo, B2 microglobulina normal, marcadores tumorales negativos, los estudios reumatológicos mostraron: ANA resultado positivo, con descripción patrón mixto: patrón nuclear moteado, título 1/320. Patrón nuclear moteado con puntos aislados>de 10. Anti-ENA screen IGG (rnp, sm, ss-) 130,31 (positivo), autoanticuerpo anti-RNP/SM 9,05 (negativo), autoanticuerpo anti smith sm 2,86 (negativo), anti-SSa (ro) 164,7 (positivo), ANCA negativo, anticardiolipina (ACA) negativo, anti beta 2 glicoproteína positivo, COOMBS directo positivo, anticoagulante lúpico positivo, C3 - C4 negativo y anti-DNA-DS negativo. Proteinuria de 24 h 500mg/24h, con proteinograma electroforético sérico con hipoproteinemia y en orina con hipoproteinemia no significativa. Líquido cefalorraquídeo con citoquímica glucosa 53mg/dl, proteínas 218mg/dl, celularidad 0, gram negativo, cultivo negativo, PAP negativo, citometría de flujo negativo.
Se complementó estudios con electromiografía: estudio neurofisiológico evidencia polineuropatía subaguda crónica mixta primariamente desmielinizante predominante sensitiva, simétrica, distal que afecta cara y cuatro extremidades con mayor severidad en MMII. Por último, se realizó biopsia de glándula salival: con resultado de Focus Score: 1. Estos resultados fueron compatibles con nuestra hipótesis, se debe descartar aquellas patologías que pueden cursar con polineuropatías principalmente colagenopatías, gammapatías o paraneoplásicos, ya que el tratamiento difiere según la etiología de la polineuropatía.
Se inició pulsos de metilprednisolona 250mg c/24h durante 2 días, luego se indica prednisona de 50mg c/24h vía oral, con todos los resultados fue evaluada por reumatología por la impresión diagnóstica de SS con compromiso del sistema nervioso periférico, el cual indicó iniciar infusión con ciclofosfamida 750mg en 3 h, el paciente presentó leve mejoría clínica, continúa en seguimiento por reumatología.
Caso 2Paciente mujer de 75 años, con antecedente de hipertensión arterial, hipotiroidismo, glaucoma y ojo seco. Con una duración de la enfermedad de 2 años y 6 meses iniciando con parestesia y disestesia en planta de pie derecho, tras 1 mes los síntomas se extendieron a miembro superior derecho, después de 2 meses se agregaron hacia hemicuerpo izquierdo, los cuales progresaron gradualmente, originando dificultades para la deambulación que requirió uso de andador. La paciente estuvo siendo evaluada por consultorio externo de forma irregular.
Al examen físico Glasgow 15/15, se presenta despierta, alerta, atenta, orientada, movimientos pseudoatetósicos en miembros superiores, debilidad distal global a predominio derecho, arreflexia global, alteración de propiocepción severa, sin signos meníngeos.
Durante su hospitalización se planteó neuronopatía sensitiva como posibilidad diagnóstica, secundaria a probable SS paraneoplásico o primaria. Dado los síntomas SICCA que precede a la paciente se planteó como primera posibilidad SS, se solicitaron exámenes de laboratorio como hemograma, electrólitos y bioquímico normales, perfil hepático normal, perfil tiroideo normal, vitamina B12 y ácido fólico normal, VSG normal, LDH normal, HBA1c normal, B2 microglobulina normal, TORCH negativo, VIH negativo, HLTV negativo, VHB-VHC negativo, marcadores tumorales negativos, ANA 1/100, 1/320, ENA 4.86, ANCA negativo, factor reumatoideo normal, ACA, anti beta 2 glicoproteína y anticoagulante lúpico (AL) negativo.
Se realizó estudios de imágenes como tomografía axial computarizada de tórax, abdomen y pelvis con contraste sin alteraciones, PET SCAN negativo. En la electromiografía se encontró ganglionopatía sensitiva. Signos de reinervación de distribución asimétrica a predominio de la extremidad inferior derecha, sugerentes de neuronopatía motora. Patrón de interferencia inconstante por el gran compromiso sensitivo. Por último, se realizó biopsia de glándula salival con resultado de focus score 2.
Respecto al paciente con ganglionopatía sensitiva asociada a SS primario se decide usar infusión de ciclofosfamida 750mg en 2 h, la paciente presenta leve mejoría clínica, aún sigue en controles ambulatorios a cargo de reumatología.
Caso 3Paciente mujer de 51 años con antecedente médico de ojo seco de 5 años de evolución. Con duración de la enfermedad de 6 meses, inicialmente caracterizado por parestesias en región abdominal baja, el cual aumentó gradualmente, extendiéndose hacia nivel de ambos glúteos, descendió hasta muslos y finalmente hasta piernas. La paciente estuvo siendo evaluada por consultorio externo con tratamiento analgésico y estaban pendientes exámenes de laboratorio e imágenes. Al agregarse limitación a la deambulación acude a nuestra institución.
Al examen físico se encuentra en Glasgow 15/15, despierta, alerta, atenta, nivel sensitivo desde D10, leve paraparesia asimétrica distal, reflejo plantar indiferente bilateral, marcha con cadencia aumentada. Se plantea mielopatía dorsal crónica progresiva solicitando resonancia magnética de médula dorso lumbar, donde se evidenció lesión hiperintensa a nivel de D9-D12 en T2 e hiperintensa en T1 con contraste (fig. 1). Ante la sospecha de mielitis longitudinalmente extensa se decidió iniciar metilprednisolona 1 g/día durante 5 días.
 Resonancia magnética de médula dorso-lumbar en el corte sagital en T2 y T1 con contraste, se evidencia lesión hiperintensa a nivel de D9-D12. c y d) Cortes axiales en T2 y T1 con contraste donde se objetiva captación homogénea de contraste.")
Ingresa a hospitalización para completar estudios etiológicos, fue evaluada por oftalmología el cual objetivó xerostomía, se realizó exámenes de laboratorio, los cuales son los siguientes: hemograma normal, electrólitos normales, perfil bioquímico normal, TGO 75 U/l y TGP 120 U/l, perfil lipídico normal, vitamina B12 y ácido fólico normal, perfil tiroideo normal, HIV y HTLV negativo, VHB y VHC negativo, TORCH negativo, marcadores tumorales negativo, B2 microglobulina negativo, ANA patrón nuclear moteado 1/80, complemento c3-c4 normal. Líquido cefalorraquídeo: glucosa 90mg/dl, proteínas 21,2mg/dl, recuento celular 0, Gram negativo, tinta china negativo, ADA negativo, BK y cultivo negativo, VDRL negativo, PAP negativo. Anti-MOG y anti-AQP4 en muestra sérica con método de enzimoinmunoanalisis fue negativo. El cual no fue repetido ante posibilidad alejada de enfermedad desmielinizante, debido que al ser una mielopatía crónica progresiva se consideró otros diagnósticos. Tomografía axial computarizada de tórax, abdomen y pelvis con contraste sin alteraciones. Fue evaluada por neurocirugía quien describió cuadro tumoral alejado. Ante la sospecha de SS se realizó biopsia de glándula salival con focus score: 1.
Completó pulsos de metilprednisolona y posterior a ello recibió tratamiento con prednisona 50mg c/24h vía oral, se evidenció mejora de fuerza en miembros inferiores, la cual le permitía deambular mejor. Se concretó transferencia a reumatología para manejo de manifestación neurológica de SS.
Caso 4Paciente mujer de 60 años, con antecedente no preciso de esclerodermia, enfermedad de Raynaud, artrosis en mano y columna, se presenta con un tiempo de enfermedad de 8 días caracterizado por dolor dorsolumbar en faja, fluctuante, disminución de fuerza y sensibilidad de miembros inferiores progresivo, lo que la limita y hasta impide la deambulación. Adicionalmente refiere historia de año y medio de artralgias en manos, frialdad acral, signos de Raynaud, xerostomía, xeroftalmía, disfagia ocasional con pérdida de peso de aproximadamente 5kg en 1 año.
Al examen físico presencia de lesiones maculares hipercrómicas con lesiones satélites hipocrómicas en región cervicodorsal posterior; presencia en manos de esclerodactilia, nódulos de Bouchart y Heberden; al examen neurológico Glasgow 15/15, despierta, atenta, lenguaje espontáneo, paraparesia 4/5, clonus en miembros inferiores a predominio derecho, nivel sensitivo D7, hiperreflexia, sin signos meníngeos, marcha parética con aumento de base de sustentación.
Se planteó mielopatía dorsal aguda, solicitando resonancia de columna cervical dorsal contrastada, en la cual se evidenció hiperintensidad intramedular extensa que compromete los segmentos medulares desde el nivel C5 hasta D6, de localización central con realce periférico a la administración de contraste (fig. 2). Con este resultado se inició los estudios de mielitis longitudinalmente extensa, planteando como primera posibilidad proceso inflamatorio ya sea primario del SNC (neuromielitis óptica o anti-MOG) o secundario a enfermedad sistema (SS, LES, sarcoidosis, entre otros).
 Secuencias T2 y T1 con contraste sagital donde se evidencia hiperintensidad intramedular extensa que compromete los segmentos medulares desde el nivel C5 hasta D6, de localización central con realce periférico a la administración de contraste. c y d) Imágenes axiales donde se observa hiperintensidad en T2 que compromete todo el canal medular e imagen captadora de contraste de forma homogénea.")
Resonancia magnética de médula cérvico-dorsal. a y b) Secuencias T2 y T1 con contraste sagital donde se evidencia hiperintensidad intramedular extensa que compromete los segmentos medulares desde el nivel C5 hasta D6, de localización central con realce periférico a la administración de contraste. c y d) Imágenes axiales donde se observa hiperintensidad en T2 que compromete todo el canal medular e imagen captadora de contraste de forma homogénea.
Dentro de sus exámenes complementarios presenta hemograma normal, perfil bioquímico normal, electrólitos normales, VHB-VHC negativo, TORCH negativo, Epstein Barr negativo, VDRL negativo, ANA negativo, ANCA negativo, anti beta 2 glicoproteína negativa, ACA negativo, marcadores tumorales negativos, el paciente no contó con los recursos económicos para el estudio de AQP4 y anti-MOG, en el estudio del líquido cefalorraquídeo: glucosa 75mg/dl, proteínas 48mg/dl, celularidad 1 cel/mm3, gram y cultivo negativo, VDRL negativo, tinta china y criptolátex negativo; en los estudios de imágenes TAC cuello, abdomen, tórax y pelvis contrastada sin evidencia de lesiones neoplásicas sólidas, estudio de potencial evocado visual normal.
En primera instancia por mielitis longitudinalmente extensa se decide iniciar tratamiento con pulsos de metilprednisolona; sin embargo, debido a que el uso de corticoides representa un riesgo de crisis renal esclerodérmica, se decide inicio de tratamiento con recambio plasmático por progresión de paraparesia, con lo que se evidenció respuesta lenta favorable. En curso de estudio y tratamiento se obtuvo resultado de anti-SS-A(anti-Ro) 34,7 (positivo) y anti-SS-A52 kDa 68,8 (positivo), con lo que el paciente es referido al servicio de reumatología, donde deciden iniciar tratamiento con infusión de ciclofosfamida obteniendo respuesta favorable en los controles posteriores.
DiscusiónLa prevalencia de manifestaciones neurológicas del SS en la mayoría de los estudios es de aproximadamente el 20%. La población más afectada fueron las mujeres, tal como se observa en nuestros 4 casos. El promedio de edad de afectación es de 51 +/- 12 años. En nuestros pacientes tenemos de 35-75 años, al igual que en la literatura revisada5. Hay múltiples hipótesis sobre la patogenia de las manifestaciones neurológicas, como vasculopatía de vasos pequeños, ganglionitis de la raíz dorsal, crioglobulinemia, desmielinización y disfunción autonómica mediada por anticuerpos. La variabilidad de mecanismos complica el adecuado tratamiento6.
La afectación del SNP es la más frecuente, dentro de ellas la principal es la neuropatía sensorial pura, seguida por neuropatía sensoriomotor, menos frecuente neuronopatía, afectación de nervios craneales y polirradiculoneuropatía. En el caso de nuestro primer paciente fue una polineuropatía sensitivo motora acorde a los estudios previos, a diferencia del segundo caso clínico que es una ganglionopatía el cual es una manifestación menos frecuente. La afectación del SNC más frecuente viene desde crisis epilépticas, vasculitis, meningitis, encefalitis, mielitis transversa y casos más raros de mielitis longitudinalmente extensa. En el tercer y cuarto caso presentaron mielitis longitudinalmente extensa lo cual es una manifestación poco frecuente7.
La evolución clínica tiene un curso subagudo y crónico. El promedio de tiempo de inicio de síntomas SICCA (xerostomía y xeroftalmia) y el diagnóstico de SS fue de 7,7 años (+/- 5 años)8. En nuestro primer paciente tuvo 6 meses de xeroftalmia como antecedente hasta antes del diagnóstico de SS, en el segundo caso tenía xeroftalmia de tiempo no definido, nuestro 3.er caso con xeroftalmia 5 años y el 4.° caso 1 año y medio, los cuales se asocian con la literatura presentada, excepto el primero que presentó una evolución más rápida.
El diagnóstico de SS se realiza según los criterios del Consenso del Grupo Americano Europeo. 1) Presencia subjetiva de sequedad ocular; 2) presencia subjetiva de sequedad bucal; 3) medidas objetivas de sequedad ocular por prueba de Schirmer o tinción corneal; 4) puntaje de enfoque mayor igual a 1 en un biopsia de glándula salival; 5) gammagrafía salival que muestra flujo salival reducido (1,5ml en 15 min) y/o sialectasias difusas y 6) autoanticuerpos positivos contra SS-A y/o SS-B. SS se diagnostica cuando 4 de 6 ítems están presentes; siempre que la anatomopatología de las glándulas salivales o la presencia de autoanticuerpos contra SS-A/SS-B sea positiva9. Como en el caso de nuestros pacientes en el primero se le diagnosticó xeroftalmia, ANA positivo, anti-SSA positivo y biopsia con focus score 1. En nuestro segundo caso encontramos xeroftalmia y biopsia de glándula positiva con ANA positivo y ENA positivo, en el 3.er caso presentó xeroftalmia, ANA 1/80, biopsia con focus score 1. Por último, en nuestro 4.° caso se encontró xerostomía, xeroftalmia y anti-SSA positivo.
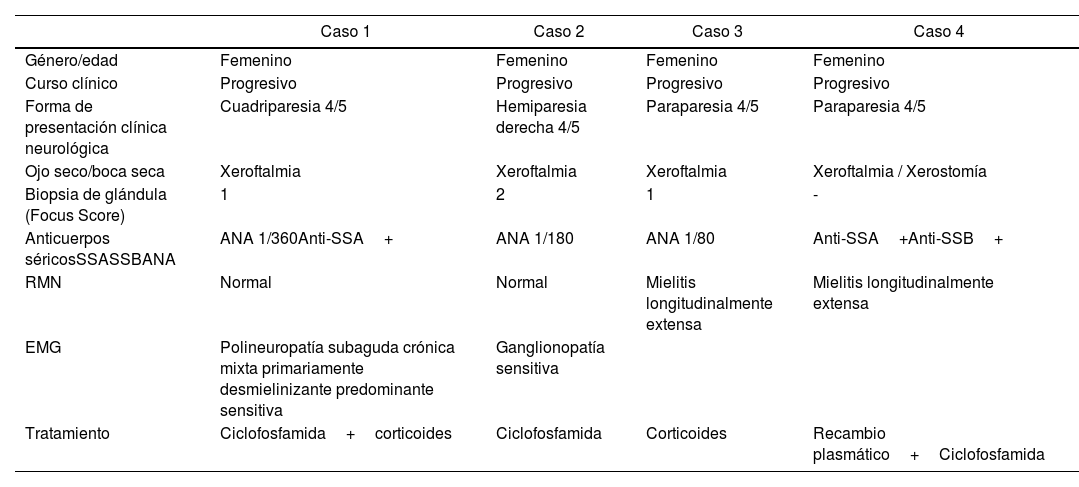
Actualmente no hay un consenso sobre el tratamiento de SS, pero los diversos estudios indican que el tratamiento inmunosupresor es de elección y se basa en la alta actividad de la enfermedad, lesiones rápidamente progresivas y síntomas severos. El uso de corticoides y ciclofosfamida endovenosa es a menudo recomendado10. Nuestro primer paciente recibió pulsos de metilprednisolona y posteriormente 2 ciclos de ciclofosfamida, en nuestro segundo paciente también recibió ciclos de ciclofosfamida, el tercero recibió pulsos de metilprednisolona durante 5 días y quedó con corticoides hasta evaluación ambulatoria, por último al cuarto caso se le inició ciclofosfamida, nuestros 4 pacientes tuvieron una favorable evolución clínica, actualmente siguen en seguimiento. Se puede mostrar un resumen de las principales características de los casos reportados en la tabla 1.
Características clínicas de los pacientes con manifestaciones neurológicas asociadas al síndrome de Sjogren
| Caso 1 | Caso 2 | Caso 3 | Caso 4 | |
|---|---|---|---|---|
| Género/edad | Femenino | Femenino | Femenino | Femenino |
| Curso clínico | Progresivo | Progresivo | Progresivo | Progresivo |
| Forma de presentación clínica neurológica | Cuadriparesia 4/5 | Hemiparesia derecha 4/5 | Paraparesia 4/5 | Paraparesia 4/5 |
| Ojo seco/boca seca | Xeroftalmia | Xeroftalmia | Xeroftalmia | Xeroftalmia / Xerostomía |
| Biopsia de glándula (Focus Score) | 1 | 2 | 1 | - |
| Anticuerpos séricosSSASSBANA | ANA 1/360Anti-SSA+ | ANA 1/180 | ANA 1/80 | Anti-SSA+Anti-SSB+ |
| RMN | Normal | Normal | Mielitis longitudinalmente extensa | Mielitis longitudinalmente extensa |
| EMG | Polineuropatía subaguda crónica mixta primariamente desmielinizante predominante sensitiva | Ganglionopatía sensitiva | ||
| Tratamiento | Ciclofosfamida+corticoides | Ciclofosfamida | Corticoides | Recambio plasmático+Ciclofosfamida |
ANA: anticuerpo antinuclear; EMG, electromiografía; RMN: imágenes por resonancia magnética; SSA: anticuerpo anti-Ro/SSA; SSB: anticuerpo anti-La/SSB.
Se presentan 4 casos de SS con compromiso del sistema nervioso tanto periférico como central, como se pudo mostrar algunas manifestaciones neurológicas pueden presentarse como el síntoma inicial de SS y también aparecer durante la evolución de los síntomas SICCA, es importante tener en cuenta los criterios diagnósticos de esta entidad, considerarla en nuestro diferenciales, los cual nos permitirá brindar un correcto tratamiento y seguimiento.
Autoría/colaboradoresTodos los autores han participado en la concepción y diseño del artículo, recolección de datos, redacción, revisión crítica del artículo y aprobación de la versión final.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
