











La enfermedad de Creutzfeldt-Jakob (ECJ) es una encefalopatía espongiforme transmisible, debido al acúmulo de una isoforma alterada de la proteína priónica (PrPsc) en el encéfalo. El cuadro clínico consiste en demencia rápidamente progresiva, ataxia y mioclonías, con alteraciones en el electroencefalograma (EEG), proteína 14-3-3 positiva en líquido cefalorraquídeo y alteraciones en resonancia magnética (RM). Su diagnóstico definitivo es anatomopatológico.
Caso clínicoMujer de 64 años con ataxia rápidamente progresiva, trastornos visuales, disartria, disdiadococinesia dismetría y distonía axial. Apraxia, amiotrofia mioclonías y crisis convulsivas. Fallece a los 4 meses de inicio con marcado deterioro motor, rigidez y postura en flexión, con escaso deterioro cognitivo conductual. Se realiza estudio anatomopatológico con biología molecular.
ConclusionesPresentamos un caso de ECJ «definida» con clínica infrecuente de ataxia, mioclonías, sin deterioro cognitivo para su genotipo (M/V1). Aún con EEG atípico y proteína 14-3-3 no concluyente, nuestra sospecha diagnóstica fue guiada principalmente por la rápida velocidad de instalación de los síntomas. En este caso nos ha resultado muy importante el estudio mediante RM encefálica de alto campo, con las secuencias adecuadas, para demostrar imágenes típicas y arribar al diagnóstico definido.
Creutzfeldt-Jakob disease (CJD) is a transmissible spongiform encephalopathy due to the accumulation of altered prion protein isoform (PrPsc) in the brain. The typical clinical symptoms are rapidly progressive dementia, ataxia, myoclonus, electroencephalogram (EEG) alterations, with positive 14-3-3 protein in cerebrospinal fluid and changes in magnetic resonance imaging (MRI). The definitive diagnosis is done anatomopathologically.
Case reportA 64 year old female patient who presented with rapid progressive ataxia, visual disturbances, dysarthria, dysdiadochokinesia, dysmetria and axial dystonia. She also presented with apraxia, atrophy, myoclonus and seizures. She died 4 months after onset, with severe motor deterioration, rigidity, flexed posture and low cognitive-behavioral impairment. We did an anatomopathological study in addition to molecular biology.
ConclusionsWe present a “defined” case study of ECJ with infrequent symptoms of ataxia, myoclonus, but with no cognitive deterioration for its genotype (M/V1). Despite inconclusive 14-3-3 protein levels and atypical EEG, our diagnosis was mainly guided by the rapid onset of symptoms, in which high-field MRI (with the appropriate sequences) were key in showing typical images and in defining the diagnosis.
La enfermedad de Creutzfeldt-Jakob (ECJ)4–6 es una encefalopatía espongiforme transmisible, debido al acúmulo de una isoforma alterada de la proteína priónica (PrPsc) en el encéfalo. El cuadro clínico característico consiste en demencia rápidamente progresiva, ataxia y mioclonías, con descargas sincrónicas seudoperiódicas en el electroencefalograma (EEG), proteína 14-3-3 positiva en líquido cefalorraquídeo (LCR) y señal hiperintensa en ganglios basales y/o corteza cerebral (tablas 1 y 2).
Criterios diagnósticos para la vigilancia de la ECJ de la OMS
| I.1 Definida: confirmada por neuropatología/inmunohistoquímica |
| I.2 Probable: I+2 de ii+iii o iv Posible+14-3-3 positivo |
| I.3 Posible: I+2 de ii+duración menor a 2 años |
| I. Demencia rápidamente progresiva. |
| II. Mioclonías Trastornos visuales o cerebelosos Signos piramidales o extrapiramidales Mutismo acinético |
| III. EEG típico |
| IV. Señal hiperintensa en núcleo caudado/putamen en RM encefálica |
Se resaltan en negritas los datos positivos en el caso clínico presentado.
Tomado de EuroCJD1.
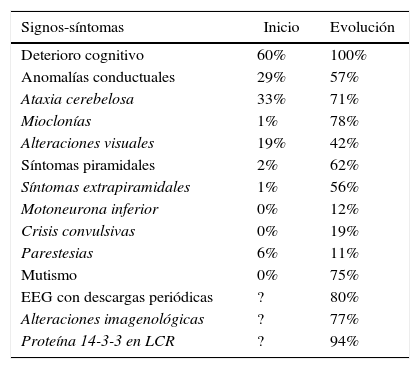
Características clínicas de la ECJ presentes al inicio o durante la evolución de la enfermedad
| Signos-síntomas | Inicio | Evolución |
|---|---|---|
| Deterioro cognitivo | 60% | 100% |
| Anomalías conductuales | 29% | 57% |
| Ataxia cerebelosa | 33% | 71% |
| Mioclonías | 1% | 78% |
| Alteraciones visuales | 19% | 42% |
| Síntomas piramidales | 2% | 62% |
| Síntomas extrapiramidales | 1% | 56% |
| Motoneurona inferior | 0% | 12% |
| Crisis convulsivas | 0% | 19% |
| Parestesias | 6% | 11% |
| Mutismo | 0% | 75% |
| EEG con descargas periódicas | ? | 80% |
| Alteraciones imagenológicas | ? | 77% |
| Proteína 14-3-3 en LCR | ? | 94% |
Desde el punto de vista neuropatológico, la ECJ esporádica se caracteriza por degeneración espongiforme, pérdida neuronal, gliosis y presencia de agregados de formas anormales de la proteína priónica (PrP), que son resistentes a la proteasa.
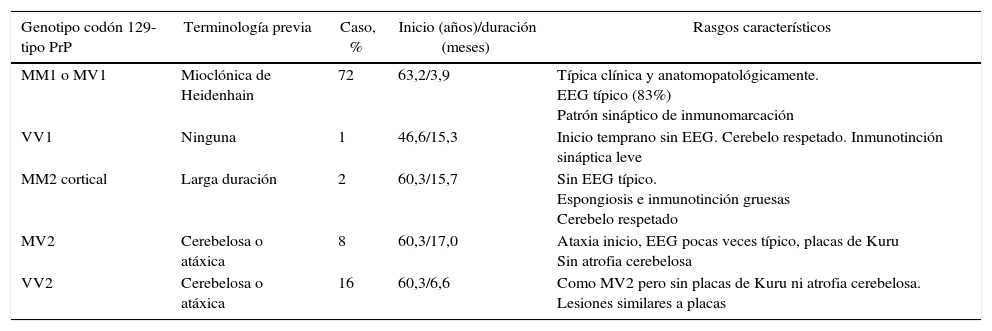
A pesar de estas características descriptas y conocidas, la enfermedad muestra una amplia variabilidad fenotípica, que ha permitido la identificación y la caracterización de subtipos clinicopatológicos, definidos de acuerdo también con su base molecular (tabla 3).
Clasificación de la ECJ esporádica
| Genotipo codón 129- tipo PrP | Terminología previa | Caso, % | Inicio (años)/duración (meses) | Rasgos característicos |
|---|---|---|---|---|
| MM1 o MV1 | Mioclónica de Heidenhain | 72 | 63,2/3,9 | Típica clínica y anatomopatológicamente. EEG típico (83%) Patrón sináptico de inmunomarcación |
| VV1 | Ninguna | 1 | 46,6/15,3 | Inicio temprano sin EEG. Cerebelo respetado. Inmunotinción sináptica leve |
| MM2 cortical | Larga duración | 2 | 60,3/15,7 | Sin EEG típico. Espongiosis e inmunotinción gruesas Cerebelo respetado |
| MV2 | Cerebelosa o atáxica | 8 | 60,3/17,0 | Ataxia inicio, EEG pocas veces típico, placas de Kuru Sin atrofia cerebelosa |
| VV2 | Cerebelosa o atáxica | 16 | 60,3/6,6 | Como MV2 pero sin placas de Kuru ni atrofia cerebelosa. Lesiones similares a placas |
Tomado de Parchi et al.3.
En nuestro caso el genotipo obtenido de la autopsia no es congruente con los rasgos clínicos descriptos característicamente para el mismo.
Caso clínicoSe presenta una mujer de 64 años, de profesión repostera, que consulta por un cuadro clínico de ataxia progresiva de 2 meses de evolución.
Antecedente personal: hipertensión arterial medicada con bisoprolol.
Antecedente familiar: padre fallecido a los 55 años, con síntomas cognitivos y motores de rápida evolución, sin diagnóstico.
Inicia con ataxia rápidamente progresiva de la marcha y tronco, trastornos visuales corticales y oculomotores (heminegligencia, nistagmo horizontal, alteraciones de la mirada conjugada y en los movimientos de seguimiento ocular lentos), disartria (palabra escandida), disdiadococinesia, dismetría y distonía axial. Se constata además apraxia (mano ajena derecha), amiotrofia y mioclonías espontáneas, de acción y reflejas, de inicio hemicorporal derecho con posterior generalización.
En las siguientes 4 semanas progresa con acentuación sintomática, aumento de las mioclonías que le impiden higienizarse, alimentarse, ataxia invalidante con imposibilidad de mantenerse sentada, falta de sostén cefálico y alteración deglutoria. Agrega crisis convulsivas tónico-clónicas generalizadas, de difícil control.
Fallece a los 4 meses de inicio sintomático, por intercurrencia infecciosa, con marcado deterioro motor, rigidez y postura en flexión, lúcida, orientada, con escaso deterioro cognitivo-conductual. Se realiza autopsia cerebral para evaluación anatomopatológica y biología molecular.
Estudiada en su inicio en otro hospital, donde se habían realizado exámenes analíticos exhaustivos (hemograma, velocidad de eritrosedimentación, bioquímica, serología, autoanticuerpos, anticuerpos tumorales, dosaje de tóxicos, neurotransmisores y vitaminas), bioquímica y cultivo de LCR. Estudios imagenológicos: tomografía computarizada de tórax, abdomen y pelvis, ecografía Doppler de vasos del cuello, senografía, ecografía tiroidea y ginecológica. Fondo de ojo y electromiografía con velocidad de conducción motora de 4 miembros, todos con resultados normales.
Videonistagmografía, con upbeat nistagmo en pruebas posturales.
RM encefálica (0,5 tesla) (6 de junio del 2012): normal (fig. 1), y columna dorsolumbar: procesos espondiloartrósicos.
 (0,5 tesla). Sin lesiones específicas.")
RMI encefálica (1,5 tesla) (19 de junio del 2012): moderada retracción de la corteza cerebral, en sustancia blanca subcortical pequeños focos de hiperintensidad, puntiformes (frontal precentral derecho y parietal izquierdo), hiperintensos en T2 y FLAIR e hipointensos en T1, sin modificaciones posteriores al contraste (fig. 2).
 (1,5 tesla). Moderada retracción de la corteza cerebral, en sustancia blanca subcortical pequeños focos de hiperintensidad, puntiformes (frontal precentral derecho y parietal izquierdo), hiperintensos en T2 FLAIR")
Columna cervical: procesos de espondilodiscoartrosis de C3 a C7. Estenosis de canal cervical, sin mielopatía.
Al recibir el caso realizamos un EEG con fotoestimulación: lentificación de la actividad basal, sin patrón de descargas periódicas.
Se repite una punción lumbar para la determinación en el LCR de proteína 14-3-3 por Western-Blot (técnica con sensibilidad del 94% y especificidad del 84%): resultado positivo tenue (0,5/5 en escala arbitraria de 5/5) 25 días antes del fallecimiento.
Solicitamos neuroimágenes de alto campo, RM encefálica (3,0 tesla) (2 de agosto del 2012), a 45 días de la primera, secuencias de T2 FLAIR (fig. 3), difusión (fig. 4) y tensor de difusión (factor de isotropía) (fig. 5): lesiones hiperintensas en el efecto T2 FLAIR y difusión, especialmente en la sustancia gris, la corteza temporal, el girus angular izquierdo, los núcleos ventrales posteriores del tálamo, la cabeza de los núcleos caudados y la corteza cerebelosa. Sustancia blanca notablemente preservada.
 (3,0 tesla). Lesiones hiperintensas en el efecto T2 FLAIR. Puede observarse que la sustancia blanca está notablemente preservada y la patología predomina en la sustancia gris.")
 (3,0 tesla). Lesiones hiperintensas, especialmente en la sustancia gris, la corteza temporal, el girus angular izquierdo, los núcleos ventrales posteriores del tálamo, la cabeza de los núcleos caudados y la corteza cerebelosa.")
 (3,0 tesla). Aumento de la isotropía, a expensas de una disminución de la celularidad en el coeficiente de difusión.")
Autopsia: estudio histopatológico: cambios espongiformes (++++) en todas las capas de la corteza cerebral, solo focalmente confluyentes. Inmunomarcación para GFAP: extensa hiperplasia astroglial reactiva en relación con las vacuolas. Inmunomarcación para PrP: positiva sináptica y confluente en todo el espesor de la corteza.
Biología molecular: caracterización del codón polimórfico 129 del gen PRNP (metionina/valina) mediante enzima de restricción (RFLP). Resultado genotipo (M/V) en codón 129 (tabla 3; fig. 6).
 del gen PrnP mediante una enzima de restricción (RFLP). MV en codón 129 del gen del prion humano. Polimorfismo M/V 129 del gen PRNP.ATG: metionina; GTG: valina.")
Western Blot PrP postratamiento con proteasa K, resultado positivo (+) tipo 1. Clasificación actual: ECJ definida.
ComentariosAún en ausencia de deterioro cognitivo, requerimiento indispensable para cumplir con criterios de ECJ «posible» y «probable», para arribar al diagnóstico «definido», en este caso, fue importante la sospecha clínica basada en la rápida evolución y la duda sobre el antecedente paterno.
Los métodos complementarios aportaron información inespecífica, en estadios clínicos tempranos.
Con la repetición de neuroimágenes, aplicando técnicas de mayor complejidad y especificidad (RM de alto campo, secuencias T2 FLAIR, difusión y tensor de difusión), se pudo demostrar lesiones características, acordes con el rápido deterioro clínico. Esta situación nos resultó fundamental para decidir la práctica de la autopsia cerebral, la que finalmente, por estudios histológicos y de biología molecular, permitió arribar al diagnóstico de ECJ definida, sin haber reunido los criterios previos de posible y probable según la Organización Mundial de la Salud (tabla 1).
El fenotipo de presentación atáxica-mioclónica, como en este caso, solo está descripto en 1/3 de los pacientes con ECJ esporádica con genotipo M/V1.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A Jorge Nagel, especialista en neuroimágenes, director de Departamento de Neurociencias, Grupo Gamma, Rosario, Santa Fe; Ana Lía Taratuto, anatomopatóloga, Departamento de Neuropatología, FLENI, Ciudad Autónoma de Buenos Aires; Christian Begue, biólogo molecular, Departamento de Neuropatología, FLENI, Ciudad Autónoma de Buenos Aires.
