




La neuromielitis óptica (NMO) es una enfermedad desmielinizante del sistema nervioso central. El descubrimiento del anticuerpo anti-AQP4 amplió el espectro a formas limitadas (NMOSD). El objetivo es describir los hallazgos clínicos, de laboratorio, neuroimágenes y tratamiento en estos grupos de pacientes (NMO y NMOSD).
Material y métodosRevisamos 15 historias clínicas, de forma retrospectiva, de pacientes con NMO/NMOSD en 2 centros de la Ciudad de Buenos Aires desde el periodo comprendido entre 2009 a 2013.
ResultadosGrupo 1. NMO definida (n: 7), sexo femenino: 6/7, edad: mediana: 34 años, anti-AQP4 positivo: 2/5, negativo: 3/5, no se realizó: 2/7, BOC negativas: 7/7. Primer brote: NO: 5, MTA: 2, curso clínico recidivante: 4/7, monofásico: 3/7, RM de encéfalo: normal: 2/7, lesiones inespecíficas: 5/7. RM espinal: MTA longitudinal extensa (LETM): 7/7. Tratamiento agudo: metilprednisolona: 7/7; preventivo: azatioprina más meprednisona: 7/7.
Grupo 2. NMOSD limitada (n: 8) (NO recurrente [n: 3], NO aislada [n: 3] y MTA aislada [n: 2]) sexo femenino: 7/8, edad: mediana: 30 años, anti-AQP4 positivo: 8/8, BOC positivas: 2/8. RM de encéfalo: normal: 2/8, lesiones inespecíficas 5/8. RM de columna: LETM: 2/8. Tratamiento agudo: metilprednisolona: 8/8; preventivo: azatioprina más meprednisona: 8/8.
ConclusionesLa NMO/NMOSD se observó en mujeres jóvenes y la manifestación de inicio más frecuente fue NO unilateral. Los estudios diagnósticos más relevantes fueron la RM espinal con la presencia de mielitis longitudinal extensa en todos los casos de NMO definida y en las mielitis aisladas, y los anticuerpos anti-AQP4 principalmente en las formas limitadas, donde por definición fueron positivos en un 100% de los casos.
The neuromyelitis optica (NMO) is a demyelinating disease of central nervous system central. Discovery of anti-AQP4 broadened the spectrum to limited forms (NMOSD). The objective is to describe the clinical, laboratory, neuroimaging and treatment in these patient groups (NMO and NMOSD).
Material and methodsWe review 15 medical records, retrospectively, of patients with NMO/NMOSD at 2 centers in the Buenos Aires City from the period 2009 to 2013.
ResultsGroup 1: definite NMO (n=7), female: 6/7, age: median: 34 years, anti-AQP4 positive: 2/5, negative: 3/5, was not performed: 2/7, negative OCB: 7/7. First attack: NO: 5, ATM: 2, relapsing clinical course: 4/7, monophasic: 3/7, brain MRI: Normal: 2/7, nonspecific lesions: 5/7. Spinal MRI: longitudinal extensive MTA (LETM): 7/7. Acute treatment: methylprednisolone: 7/7; preventive: meprednisone assosiated with azathioprine: 7/7.
Group2: NMOSD limited (n=8) (ON recurrent (n=3), ON alone (n=3) and MTA alone (n 2)) female: 7/8, age: median: 30 years, anti - AQP4 positive: 8/8, BOC positive: 2/8. MRI brain: Normal: 2/8, nonspecific lesions 5/8. Spinal RM: LETM: 2/8. Acute treatment: methylprednisolone: 8/8; preventive: meprednisone associated an azathioprine: 8/8.
ConclusionsThe NMO/NMOSD was observed in young women and the most common manifestation of onset was unilateral ON. Diagnostic studies most relevant were spinal MRI with the presence of longitudinal extensive myelitis transverse in all cases of definite NMO and isolated myelitis and the anti-AQP4 were positive in 100% cases of limited NMO.
La neuromielitis óptica (NMO) es una enfermedad inflamatoria, desmielinizante del sistema nervioso central (SNC) caracterizada por ataques de neuritis óptica (NO) y mielitis transversa (MTA) que usualmente sigue un curso de brotes y remisiones, con acumulación de déficits neurológicos que pueden ser irreversibles, y progresión de discapacidad principalmente en pacientes adultos jóvenes1–3. Durante décadas fue considerada como un subgrupo clínico de esclerosis múltiple (EM). Sin embargo, en la actualidad existen múltiples características clínicas y paraclínicas que diferencian ambas entidades. Sin duda, lo que marcó un nuevo enfoque de la enfermedad fue el descubrimiento del autoanticuerpo IgG1 anti-aquaporina 4 (AQP4), que no solo diferencia la NMO de la EM (específico para NMO), sino que también amplió el espectro de esta entidad a formas limitadas que hoy se conocen con el nombre de desórdenes del espectro de la NMO (NMOSD, del inglés), canalopatía autoinmune por AQP4 o encefalomielitis por AQP41–4. El objetivo de nuestro trabajo es describir los hallazgos tanto clínicos, de laboratorio y de las neuroimágenes como del tratamiento en pacientes con NMO definida y NMOSD.
Material y métodosSe revisaron de la base de datos, de forma retrospectiva, 15 historias clínicas de pacientes con NMOSD evaluados en 2 centros de la ciudad de Buenos Aires (Hospital General de Agudos Dr. Carlos G. Durand y Hospital General de Agudos Dr. Teodoro Álvarez), desde el periodo de enero de 2009 a enero 2013 y se realizó una descripción de los datos obtenidos.
Este estudio consideró como criterios de inclusión a pacientes con diagnóstico NMO definida y NMOSD con AQP4 positivo. Se tomó como diagnóstico definido de NMO los criterios de Wingerchuck et al., de 20062, donde son necesarios los 2 criterios mayores: mielitis transversa aguda (MTA) más neuritis óptica (NO) y al menos 2 de los siguientes criterios: RM de encéfalo inicial normal o que no cumpla con los criterios de Barkhoff/Tintoré para EM, RM espinal con afección de al menos 3 segmentos medulares (LETM) e IgG-NMO o AQP4 positiva en suero. Definimos NMOSD según Wingerchuck et al., 20071, a las formas limitadas como la NO aislada bilateral o recurrente, LETM aislada o recurrente, EM óptico-espinal, NO o LETM asociada a enfermedades autoinmunes y NO o LETM asociada a lesiones típicas de NMO. Asimismo, incluimos datos relevantes como edad, sexo, síntomas/signos de inicio, curso clínico, datos de laboratorio para enfermedades autoinmunes, bandas oligoclonales (BOC) en líquido cefalorraquídeo (LCR), RM de encéfalo y espinal, tratamiento del brote y preventivo.
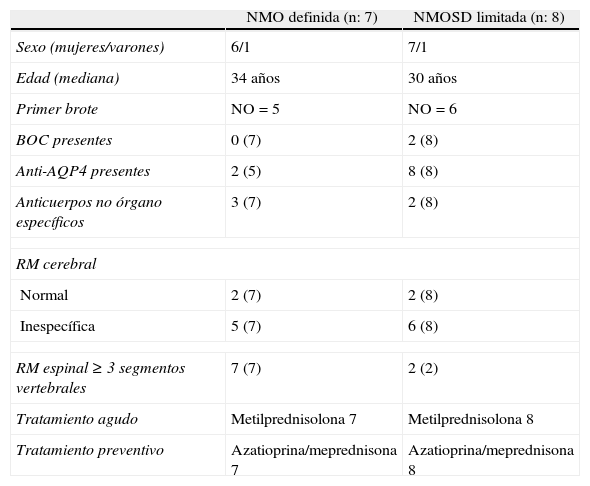
ResultadosEn el grupo 1 se evaluaron 7 pacientes que cumplían criterios de NMO definida de los cuales 6 eran de sexo femenino (6/7). Respecto a la edad encontramos una mediana de 34 años (rango: 19-58 años). Los anticuerpos anti-AQP4 por IFI confocal en corte de cerebelo de mono fueron positivos en 2 de 5 pacientes, negativos en 3 de 5 y no se realizaron en 2 de 7 (por motivos de disponibilidad, falta de recursos y por cumplir criterios). Las BOC realizadas por isoelectroenfoque fueron negativas en todos los pacientes (7/7). Con respecto al primer brote encontramos NO en 5 sujetos (todas unilaterales) y MTA en 2. El curso clínico recidivante en 4/7 de las cuales 3 fueron NO recidivante (una NO bilateral) y 2 MTA recidivante (una paciente presentó NO más MTA recurrente), curso monofásico se observó en 3 de 7 pacientes. Otros signos de interés fueron encontrados: vómitos recurrentes (2/7), Lhermitte (2/7) y espasmos tónicos paroxísticos dolorosos (4/7). Cuando evaluamos las neuroimágenes encontramos que la RM de encéfalo fue normal en 2 de 7 y lesiones inespecíficas se observaron en 5 de 7 pacientes. Dichos sujetos no cumplían criterios de Barkhoff/Tintoré para EM. Asimismo, no presentaban lesiones en sectores con alta expresión de AQP4. Ninguno de los pacientes presentó lesiones que capten contraste. La RM espinal mostró MTA longitudinal extensa (LETM) en todos los pacientes (7/7) y 4 de 7 presentaron lesiones gadolinio positivas. El sector más frecuentemente afectado fue el nivel cervicodorsal (4/7). El tratamiento agudo se realizó con metilprednisolona en todos los pacientes (7/7) y 2 de ellos requirieron plasmaféresis como tratamiento de segunda línea por pobre respuesta a los corticoides endovenosos. El tratamiento preventivo con azatioprina más meprednisona se realizó en el 100% (7/7) (tablas 1 y 2).
Características generales de la muestra
| NMO definida (n: 7) | NMOSD limitada (n: 8) | |
| Sexo (mujeres/varones) | 6/1 | 7/1 |
| Edad (mediana) | 34 años | 30 años |
| Primer brote | NO=5 | NO=6 |
| BOC presentes | 0 (7) | 2 (8) |
| Anti-AQP4 presentes | 2 (5) | 8 (8) |
| Anticuerpos no órgano específicos | 3 (7) | 2 (8) |
| RM cerebral | ||
| Normal | 2 (7) | 2 (8) |
| Inespecífica | 5 (7) | 6 (8) |
| RM espinal≥3 segmentos vertebrales | 7 (7) | 2 (2) |
| Tratamiento agudo | Metilprednisolona 7 | Metilprednisolona 8 |
| Tratamiento preventivo | Azatioprina/meprednisona 7 | Azatioprina/meprednisona 8 |
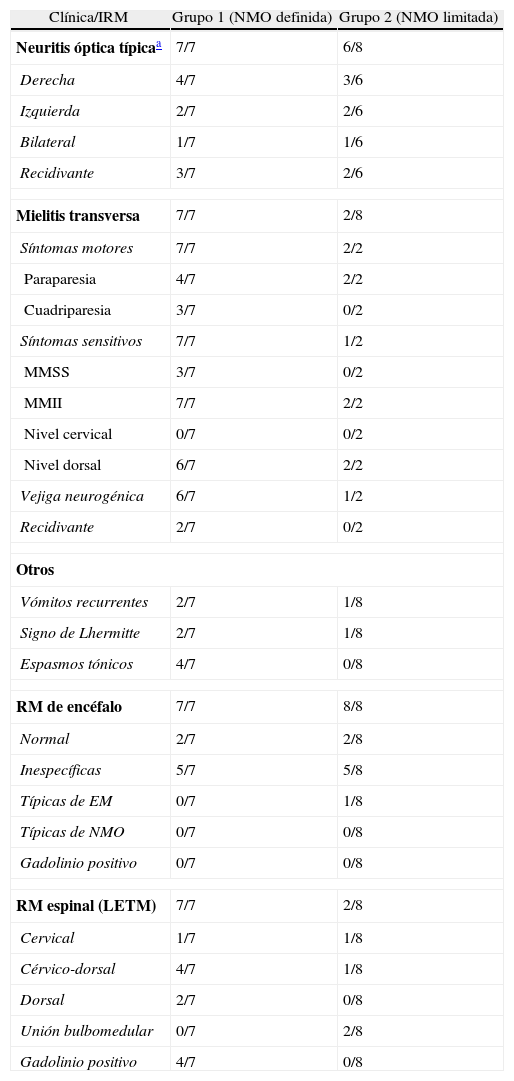
Características clínicas y neuroimágenes
| Clínica/IRM | Grupo 1 (NMO definida) | Grupo 2 (NMO limitada) |
| Neuritis óptica típicaa | 7/7 | 6/8 |
| Derecha | 4/7 | 3/6 |
| Izquierda | 2/7 | 2/6 |
| Bilateral | 1/7 | 1/6 |
| Recidivante | 3/7 | 2/6 |
| Mielitis transversa | 7/7 | 2/8 |
| Síntomas motores | 7/7 | 2/2 |
| Paraparesia | 4/7 | 2/2 |
| Cuadriparesia | 3/7 | 0/2 |
| Síntomas sensitivos | 7/7 | 1/2 |
| MMSS | 3/7 | 0/2 |
| MMII | 7/7 | 2/2 |
| Nivel cervical | 0/7 | 0/2 |
| Nivel dorsal | 6/7 | 2/2 |
| Vejiga neurogénica | 6/7 | 1/2 |
| Recidivante | 2/7 | 0/2 |
| Otros | ||
| Vómitos recurrentes | 2/7 | 1/8 |
| Signo de Lhermitte | 2/7 | 1/8 |
| Espasmos tónicos | 4/7 | 0/8 |
| RM de encéfalo | 7/7 | 8/8 |
| Normal | 2/7 | 2/8 |
| Inespecíficas | 5/7 | 5/8 |
| Típicas de EM | 0/7 | 1/8 |
| Típicas de NMO | 0/7 | 0/8 |
| Gadolinio positivo | 0/7 | 0/8 |
| RM espinal (LETM) | 7/7 | 2/8 |
| Cervical | 1/7 | 1/8 |
| Cérvico-dorsal | 4/7 | 1/8 |
| Dorsal | 2/7 | 0/8 |
| Unión bulbomedular | 0/7 | 2/8 |
| Gadolinio positivo | 4/7 | 0/8 |
EM: esclerosis múltiple; IRM: imágenes por resonancia magnética; LETM: mielitis transversa longitudinal extensa; MMII: miembros inferiores; MMSS: miembros superiores; NMO: neuromielitis óptica; RM: resonancia magnética.
En el grupo 2 encontramos 8 pacientes con NMOSD limitada, de los cuales 3 presentaban NO unilateral recurrente (un paciente asoció vómitos recurrentes), 3 NO aislada (una con afección bilateral) y 2 MTA aislada (una con signo de Lhermitte). La edad mediana fue de 30 años (rango: 21-61) y el sexo femenino fue el más frecuentemente encontrado (7/8). Los anticuerpos anti-AQP4 fueron positivos en todos los pacientes (8/8) y las BOC solo fueron positivas en 2 de 8. La RM de encéfalo fue normal en 2 sujetos y lesiones inespecíficas fueron observadas en 5 pacientes. La RM espinal mostró LETM en los 2 pacientes con MTA (100%). Es importante aclarar que ninguna lesión (encéfalo y espinal) presentó realce con el contraste en este grupo. El tratamiento agudo se realizó con metilprednisolona en todos los pacientes con buena respuesta, y de manera preventiva se utilizó azatioprina más meprednisona en todos los sujetos (8/8) (tablas 1 y 2).
DiscusiónEn nuestro estudio encontramos en ambos grupos de pacientes un franco predominio de mujeres (86,66%) jóvenes (mediana: 34 años). Estos datos avalan que la NMO y su espectro es 9 veces más prevalente en mujeres que en varones, y la edad media de inicio está entre los 30-40 años, un promedio de unos 5 años superior que en la EM tal y como refiere la literatura internacional1–3.
Cuando analizamos las características clínicas observamos que la NO unilateral fue la manifestación más frecuente del primer brote, con el 73,33% (11/15), encontrando NO derecha: 63,6%, NO izquierda: 36,4% y NO bilateral: 18,2%. Con respecto a estos resultados es importante tener en cuenta que si bien la NO bilateral es más sugestiva de NMO (en comparación con la EM) no es la forma más frecuente de presentación, y nuestros resultados se asemejan a otras publicaciones (18-30%)3,5,6. No hemos encontrado características clínicas sobresalientes que distingan las NO debidas a NMO de otras entidades. Sin embargo, no incluimos en el trabajo un seguimiento a largo plazo de las secuelas a nivel de la agudeza visual. Estudios internacionales avalan que la ceguera ocurre en el 60% de las formas recurrentes y en el 22% de las monofásicas6,7.
Respecto de la MTA encontramos que el 100% presentó clínica de trastornos motores, sensitivos y un menor porcentaje trastornos esfinterianos. El 57,15% se presentó con debilidad en los miembros inferiores (MMII) y el 42,85% además agregó debilidad uni o bilateral en los miembros superiores (MMSS) con cuadriparesia. Un dato interesante es la presencia de espasmos tónicos dolorosos en el 44,44% de los pacientes que sufrieron LETM (4/9), con un tiempo medio de 30 días posterior al ataque de MTA. Esta manifestación clínica se ha descrito con una frecuencia entre el 14% hasta el 95% en distintas series de casos8–10, por lo cual algunos investigadores han propuesto este signo como potencial criterio de apoyo para el diagnóstico de NMO9–11. El signo de Lhermitte se encontró en el 20% (33% en las series internacionales)1,6,7, el hipo y las náuseas persistentes e intratables se presentan en el 17-43% de las series europeas y norteamericanas, sin embargo en nuestra muestra el 20% presentó vómitos recurrentes y ningún paciente hipo1,6,7,12.
Lennon et al.3 en 2004 reportaron la presencia de un autoanticuerpo IgG1 específico (91%) y sensible (73%) en suero que revolucionó la forma de aproximación diagnóstica de estos pacientes, su diferenciación con la EM y además amplió el espectro a formas limitadas. Desde su descripción se han realizado múltiples trabajos con distintos sustratos y métodos presentando variaciones en la sensibilidad y la especificidad1–3,4–7,13. En nuestra muestra la frecuencia de detección del autoanticuerpo AQP4 fue del 40% en el grupo 1 (en 2 pacientes no se realizó, ya que cumplían criterios) y el 100% en el grupo 2 con una prevalencia total en ambos grupos de 76,92% (10/13). De esto se desprende que si bien la AQP4 es específica para la NMO no es imprescindible para el diagnóstico de NMO definida, pero sí lo es para la formas limitadas.
Con respecto a otros análisis bioquímicos encontramos un 33,33% (5/15) de anticuerpo antinuclear (ANA) positivo (títulos mayores de 1:160) realizado mediante IFI en líneas celulares HEp-2 con un predominio en el grupo 1 (NMO definida). En un estudio que evaluó pacientes con NMO y LETM se encontró que el 43,8% presentaba el ANA14. Las BOC fueron encontradas en un bajo porcentaje de pacientes (2/15=13,33%) que aumenta al 25% si tomamos a los pacientes del grupo 2. Datos de la literatura internacional marcan una prevalencia de 15-30% de los pacientes con NMO1.
Típicamente la RM evidencia lesiones que afectan 3 o más segmentos medulares (LETM: mielitis longitudinal extensa), generalmente cérvico-dorsales y con realce con contraste durante los brotes de MTA1–8,15. En nuestra serie el 100% de los pacientes con clínica de síndrome medular agudo presentaron LETM. Cinco (55,5%) con lesión cérvico-dorsal, 33,3% dorsal y 13,3% en la unión bulbomedular. Asimismo, el 27% de las lesiones medulares captaron contraste. Con respecto a las imágenes de encéfalo, en el trabajo de Pittock et al.16 se observó que el 60% de los pacientes tenían lesiones inespecíficas asintomáticas en el SNC, el 10% tenían lesiones en sitios con alta expresión de AQP4 (tálamo o hipotálamo, tronco o cerebelo) y el 10% cumplían criterios por imágenes de EM. En nuestra serie la RM de encéfalo al inicio fue normal en el 26,66% (4/15) y en el 76,33% (11/15) restante se encontraron lesiones inespecíficas que no cumplían criterios de EM ni se encontraban en los sitios anteriormente descritos.
El tratamiento se realizó de acuerdo con las recomendaciones de expertos, dado que no existen estudios clínicos con alto nivel de evidencia que las avalen. Todos los pacientes recibieron durante la etapa aguda (brote o ataque) pulsos de metilprednisolona (3-5 días), en los casos refractarios se usó como tratamiento más frecuente plasmaféresis (solo en 2 pacientes). Después del diagnóstico de NMO o de su forma limitada seropositiva se inició tratamiento preventivo con meprednisona más azatioprina17,18. En este último punto cabe destacar que la presencia de IgG anti-AQP4 no solo tiene valor diagnóstico, sino también pronóstico: su presencia en NO y MTA aumenta el riesgo de recurrencia y se recomienda iniciar tratamiento con inmunosupresores19–21. Es probable que el bajo porcentaje de NO recurrentes y MTA recurrentes en nuestra serie se deba al inicio temprano de inmunosupresores y al relativo corto tiempo de seguimiento.
Si bien este estudio presenta limitaciones (por su diseño retrospectivo, el número de pacientes y la falta de datos de seguimiento a largo plazo) podemos concluir que en nuestra experiencia la NMO/NMOSD se observó principalmente en mujeres jóvenes, y la manifestación de inicio más frecuente fue una NO unilateral. Los estudios diagnósticos más relevantes fueron la RM espinal con la presencia de mielitis longitudinal extensa en todos los casos de NMO definida y en las mielitis aisladas, y los anticuerpos anti-AQP4 principalmente en las formas limitadas, donde por definición fueron positivos en un 100% de los casos. Es importante tener en cuenta que durante los últimos 20 años se ha publicado extensa información sobre esta enfermedad, aunque mucho queda por investigar y descubrir21. Es por ello que creemos importante continuar con la realización de estudios en la región para entender de manera más específica el tipo de pacientes que tratamos a diario, ya que la mayoría de los datos que utilizamos son obtenidos de poblaciones con características diferentes a las nuestras.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Datos preliminares de este trabajo fueron presentados a póster (sección enfermedades desmielinizantes) en el 50 Congreso Argentino de Neurología, Mar del Plata 2013.
