






La enfermedad de Parkinson (EP) es la segunda enfermedad neurodegenerativa más común a nivel mundial en adultos mayores. Se caracteriza por la pérdida de neuronas dopaminérgicas (nDAs) en la sustancia nigra pars compacta del mesencéfalo y en algunos casos acompañada de la aparición de cuerpos intracitoplasmáticos de Lewy de α -sinucleína, signo patognomónico de la enfermedad. La EP se diagnostica clínicamente por la presencia de alteraciones motoras principalmente, y en la actualidad los tratamientos presentan nula actividad neuroprotectora. Aún no se han establecido las causas exactas de la EP, por lo que en los últimos años se ha buscado el desarrollo de modelos preclínicos más precisos, utilizando células troncales pluripotentes inducidas, permitiendo el estudio de la enfermedad de manera in vitro para generar conocimiento novedoso sobre su patogénesis y el descubrimiento de nuevos posibles blancos terapéuticos o el desarrollo de nuevos fármacos.
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease among adults worldwide. It is characterised by the death of dopaminergic neurons in the substantia nigra pars compacta and, in some cases, presence of intracytoplasmic inclusions of α-synuclein, called Lewy bodies, a pathognomonic sign of the disease. Clinical diagnosis of PD is based on the presence of motor alterations. The treatments currently available have no neuroprotective effect. The exact causes of PD are poorly understood. Therefore, more precise preclinical models have been developed in recent years that use induced pluripotent stem cells. In vitro studies can provide new information on PD pathogenesis and may help to identify new therapeutic targets or to develop new drugs.
La enfermedad de Parkinson es la segunda enfermedad neurodegenerativa con mayor incidencia a nivel mundial, en la mayoría de los casos se presenta durante la edad adulta1. Es caracterizada por 2 procesos patológicos principales: la pérdida de neuronas dopaminérgicas (nDA) en la sustancia nigra pars compacta del mesencéfalo ventral2 y la presencia de agregados intracelulares de la proteína α -sinucleína en esta misma área del cerebro, conocidos como cuerpos de Lewy3. Clínicamente se diagnostica por 4 alteraciones motoras distintivas: temblor en reposo, rigidez muscular, inestabilidad postural y bradiquinesia4. Recientemente se han identificado alteraciones no motoras relacionadas con la enfermedad, como la degeneración cognitiva, depresión, alteraciones del sueño y la pérdida del olfato5.
En la actualidad los tratamientos disponibles presentan un efecto solo sintomático, es decir, ningún fármaco tiene efecto neuroprotector en los pacientes6. El fármaco más utilizado, la levodopa, es para el control de la sintomatología motora de la enfermedad de Parkinson (EP)7 y se ha utilizado desde los años 60 del siglo xx8. La levodopa es un aminoácido que por la acción de la enzima dopadescarboxilasa en el cerebro estimula los receptores dopaminérgicos9. En cambio, las células troncales (CT) tienen el potencial de generar un modelo preclínico in vitro para enfermedades neurodegenerativas como el Parkinson., además de su alta capacidad de proliferación, su habilidad de mimetizar los diferentes estadios de la enfermedad y su relativa fácil obtención, en contraste con la obtención de tejidos post-mortem, que son regularmente utilizados para el estudio de enfermedades neurodegenerativas10.
Modelos animales utilizados en la enfermedad de ParkinsonExisten diversos modelos animales que buscan imitar la lesión neuropatológica producida en la enfermedad de Parkinson. Sin embargo, ninguno tiene la capacidad de reproducir enfermedades humanas, y es por eso que se requiere de un conjunto de modelos y técnicas para estudiar cada aspecto de la enfermedad que se manifiesta en los pacientes humanos, como la alta sensibilidad de las nDA, la formación de cuerpos de Lewy y alteraciones del movimiento10,11. Los modelos animales pueden dividirse en 2 tipos: basados en la administración de neurotoxinas y en modificaciones genéticas. Los más utilizados son aquellos basados en la administración de neurotoxinas en animales, algunas de las toxinas más estudiadas son rotenona, paraquat, 6-hidroxidopamina (6-OHDA) y 1-metil-4-fenil-1,2,3,6 tetrahidropiridina (MPTP)12–14. Adicional a estos 2 modelos principales también se utiliza la versión parcial de la vía nigrostriatal, que consiste en realizar una incisión mecánica en la ruta dopaminérgica nigrostriatal en el haz del prosencéfalo medial, resultando en una degeneración progresiva de las nDA en la SNP, mimetizando la EP15.
Dentro de los animales más utilizados para realizar el modelaje se encuentran las ratas, los ratones, el pez cebra, la mosca Drosophila melanogaster, Caenorhabditis elegans y los primates no humanos16. Modelos de la EP en Drosophila melanogaster han mostrado una disminución locomotora y dificultad para volar17. Caenorhabditis elegans disminuye su respuesta de ralentización basal, menor supervivencia, alteraciones en los ciclos de defecación y reproducción, características fenotípicas de la EP18,19. El pez cebra presenta alteraciones en su actividad locomotriz, como reducción de cruces, distancia de nado y velocidad, además de aumento del número de congelaciones y su duración20,21. Los modelos murinos sirven para estudiar la enfermedad desde un enfoque anatómico, bioquímico y conductual, debido a que brindan un manejo sencillo, con alta reproducibilidad que reflejan el estado tardío de la EP22,23. Estudios de cambios de comportamiento en ratones demuestran una disminución en coordinación, balance, función gastrointestinal, tamaño de pasos, agudeza olfatoria, dificultad para formar nidos y alteraciones en la capacidad de caminar24,25. Los modelos en ratas suelen mostrar rigidez en las extremidades, déficits cognitivos, reducción de la actividad motora, movimiento rotacional, hipocinesia, bradicinesia y asimetría postural20,26. Los primates no humanos muestran cambios conductuales análogos a los de pacientes de EP incluyendo bradicinesia, rigidez de extremidades, anormalidad postural, dificultad de mantener el balance, temblor en reposo, síndrome parkinsoniano bilateral estable, inestabilidad del gesto, deterioro de las habilidades motoras gruesas y finas16,20,27.
Modelos basados en neurotoxinasUno de los modelos con toxinas más utilizados es el de la rotenona, un plaguicida o insecticida liposoluble, que genera estrés oxidativo que daña selectivamente las neuronas dopaminérgicas, ya que inhibe el complejo i de la cadena respiratoria mitocondrial, generando deficiencias motoras características de la enfermedad12,13,28. Como modelo de la EP en ratones tiene la capacidad de reproducir las características de comportamiento de humanos y formar inclusiones intracelulares similares a los cuerpos de Lewy14,29. En ratas la exposición a rotenona genera una degeneración de las nDA y la formación de inclusiones celulares similares a los cuerpos de Lewy. Estos efectos se pueden observar como deficiencias motoras similares a la EP, incluyendo: hipocinesia, rigidez postural de manera encorvada y temblores en los miembros12,13.
Otra toxina utilizada en modelos de la EP es el paraquat o 1,1-dimetil-4,4-dipiridinio, comúnmente utilizado como herbicida, el cual produce radicales libres que reaccionan con las membranas lipídicas de las células. Se ha demostrado cierta selectividad de este compuesto hacia las nDAs de la sustancia nigra pars compacta (SNPc) positivas a tirosina hidroxilasa (TH)12,13,30. La administración sistémica en ratas del paraquat produce reducción en las actividades motoras, se reducen las fibras y neuronas TH positivas en la SNPc y presenta la capacidad de producir cuerpos de Lewy, con la desventaja de que se han obtenido resultados variables en cuanto a la muerte neuronal14.
Hasta el momento la toxina más utilizada es la 6-OHDA, presenta una actividad inhibitoria de la cadena respiratoria mitocondrial con formación de radicales libres al ser metabolizada. Tres características importantes de esta toxina son: 1) induce una degeneración rápida; 2) tiene una gran afinidad por los transportadores de noradrenalina y dopamina, provocando la muerte de neuronas adrenérgicas y dopaminérgicas; y 3) no posee la capacidad de cruzar la barrera hematoencefálica, por lo que la administración sistemática de la toxina no produce parkinsonismo, al contrario de la inyección en roedores y primates no humanos12–14,31,32. En este último, al realizar una lesión unilateral en el prosencéfalo medial se produce una pérdida de las neuronas reactivas a TH en la SNPc y una pérdida de más del 90% de nDA, generado una reducción y desbalance de las actividades motoras33,34.
En ratas la lesión unilateral con 6-OHDA produce un daño completo en las neuronas dopaminérgicas, generando un comportamiento motor asimétrico, siendo un modelo ideal para estudios de terapias de reemplazo celular y factores neuroprotectores. Por otra parte, para producir lesiones parciales es necesario disminuir la dosis en lesiones de tipo unilateral, con el propósito de estudiar los mecanismos patofisiológicos y de neurodegeneración, debido a que las lesiones causadas en el estriado causan cambios neurodegenerativos de manera progresiva en las nDA de la SNPc16.
La cuarta toxina más popular en los modelos de la EP es el MPTP, una protoxina que al ser metabolizada a través de la monoaminooxidasa B forma el metabolito 1-metil-4-fenilpiridinio (MPP+). Su mecanismo de acción se basa en la liberación excesiva de dopamina, que al ser metabolizada genera especies reactivas de oxígeno y radicales libres de manera excesiva. Adicionalmente, el MPP+es un inhibidor del complejo I de la cadena de electrones mitocondrial, disminuyendo la producción de adenosín trifosfato. Sin embargo, esta toxina no es selectiva a neuronas dopaminérgicas de la SNPc, ni suele inducir la formación de cuerpos de Lewy12,13.
Los modelos con MPTP en primates no humanos brindan información sobre posibles terapias y mecanismos patogénicos de la EP, si se realiza una lesión de manera sistémica el comportamiento será muy similar a la enfermedad presente en los humanos con EP. Sin embargo, es un procedimiento largo y con una tasa de mortalidad considerablemente alta. En cambio, los ratones exhiben características neuropatológicas y bioquímicas del daño del sistema dopaminérgico, además de una reducción en la actividad motora. Si se realiza una lesión de manera sistémica en ratones causará cierta discapacidad en el sistema dopaminérgico, ideal para estudiar procesos patofisiológicos y neurodegenerativos16. En ambas especies el MPTP causa daños en la ruta nigroestriatal con una gran pérdida de nDA en el estriado y la SNPc; la mayor desventaja es que no se ha observado la formación de cuerpos de Lewy con este modelo (tabla 1)14,35.
Comparación de diferentes modelos de la enfermedad de Parkinson con sus respectivas ventajas y desventajas
| Modelo | Tipo | Animal | Efecto | Ref. |
|---|---|---|---|---|
| Tóxico | Rotenona | Ratas | Degeneración de neuronas nigroestriadasDisminución del número de nDAFormación de cuerpos de LewyDeficiencias motoras: hipocinesia, rigidez postural y temblores en miembros | Cuenca-Alcañiz y González-Sánchez12, Blesa et al.13,Blesa et al.14 |
| Paraquat | Ratones | La aplicación sistémica reduce la actividad motora, pérdida de fibras y neuronas en SNPc, disminución de células TH positivasFormación de cuerpos de Lewy | Blesa et al.13,Blesa et al.14, Mohamed et al.44 | |
| 6-OHDA | Ratas | Produce un estado de parkinsonismo, locomoción reducidaNo se han logrado observar cuerpos de LewyLa lesión unilateral produce un daño completo en las neuronas dopaminérgicasGenera un comportamiento motor asimétricoLesiones en el estriado causan cambios neurodegenerativos de manera progresiva en las nDA de la SNPc | Blesa et al.13, Bankiewicz et al.16 | |
| Primates | Produce un estado de parkinsonismoNo se han logrado observar cuerpos de LewyLa lesión unilateral en el prosencéfalo medial produce una pérdida de las neuronas reactivas a TH en la SNPcPérdida de más del 90% de nDAReducción y desbalance de las actividades motoras | Annett et al.33, Dunnett et al.34 | ||
| MPTP | Primates | No induce la formación de cuerpos de LewyLa lesión de manera sistémica generará un comportamiento muy similar al de los humanos con EPLocomoción reducidaProcedimiento largo y con una tasa de mortalidad considerablemente altaDaños en ruta nigroestriatal con una gran pérdida de nDA en el estriado y la SNPcNo forma cuerpos de Lewy | Blesa et al.13, Blesa et al.14, Bankiewicz et al.16 Dauer y Przedborski35 | |
| Ratones | La lesión de manera sistémica causará discapacidad en el sistema dopaminérgicoReducción de la actividad motora y bradiquinesiaDaños en ruta nigroestriatal con una gran pérdida de nDA en el estriado y la SNPcNo forma cuerpos de Lewy | Blesa et al.13, Blesa et al.14, Bankiewicz et al.16 Dauer y Przedborski35 | ||
| Genético | α -syn | Ratones | Disminución de los niveles de TH y dopaminaExhibición de un comportamiento deterioradoFalta de resultados consistentes en cuanto a la neurodegeneración producida | Blesa et al.14, |
| Ratas | Se han dirigido factores virales con α -sinucleína exógena, con los cuales se ha observado enfermedad en la proteína y neurodegeneración dopaminérgica | Decressac et al.36 | ||
| LRRK2 | Ratones | Poco o nulo daño en la función de las nDA en la SNPcNeurodegeneración o cambios en la estructura neuronalKnock-out ha logrado producir acumulación de α -sinucleína y ubiquitina, inhibir la diferenciación de progenitores neuronales a nDA y aumentar la muerte celularEn el caso de una sobreexpresión del gen, se ha observado una ligera neurodegeneración de las nDA en la SNPc, pero sin alteración de los niveles de dopamina ni en la actividad locomotriz | Stoddard-Bennett T y Reijo Pera R10, Blesa et al.14, Milosevic et al.37, Hinkle et al.38, Chen et al.39 | |
| PINK1 | Ratones | Reducción gradual de los niveles de dopamina y baja actividad locomotrizNo se ha observado la formación de cuerpos de Lewy o degeneración nigroestriatal | Gispert et al.40 | |
| Drosophila melanogaster | Mutaciones en el gen PINK1 han mostrado defectos en la habilidad para volar, anormalidades en el complejo i mitocondrial derivando a niveles de ATP reducidos | Obeso et al.41, Vanhauwaert y Verstreken42 | ||
| PARKIN | Ratones | No muestra anormalidades en el comportamiento de los animales, a pesar de la ligera disminución de liberación de dopamina | Stoddard-Bennett y Reijo Pera10, Kitada et al.43 | |
| DJ-1 | Ratones | Disminución en la capacidad motoraReducción de dopamina liberada en el estriado, pero no en la SNPc | Stoddard-Bennett y Reijo Pera10, Kitada et al.43 |
α -syn: α -sinucleína; EP: enfermedad de Parkinson; MPTP: 1-metil-4-fenil-1,2,3,6-tetrahidropiridina; nDA: neuronas dopaminérgicas; 6-OHDA: 6-hidroxidopamina; SNPc: sustancia nigra pars compacta44; TH: tirosina hidroxilasa.
Es importante mencionar que los modelos de la EP basados en neurotoxinas son administrados en 3 áreas principales del cerebro: el estriado, el haz medio del prosencéfalo y la sustancia nigra (SN)11. Al ser afectadas estas regiones del cerebro se activan mecanismos compensatorios que buscan mantener la actividad neurológica, a través de alteraciones en la síntesis y liberación de dopamina, aumento en la actividad de TH, modificaciones en la actividad del estriado, el cerebelo, la corteza y las áreas corticales, creando un impacto sobre los aspectos clínicos de la EP23.
Modelos genéticosLos modelos genéticos tienen la función de simular los mecanismos relacionados con las formas genéticas de la EP. Es necesario mencionar que solo alrededor del 5% al 10% de los pacientes de la EP presentan un carácter genético4. En este caso los fenotipos patológicos y de comportamiento en los modelos murinos suelen ser diferentes a los presentados por los humanos, estudiado principalmente la función de los genes α -syn, LRRK2, PINK1, PARKIN y DJ-114. Se pueden clasificar en 3 enfoques diferentes: knock-out, sobreexpresión y transgénicos. Sin embargo, los modelos en Drosophila melanogaster, Caenorhabditis elegans y murinos no muestran las deficiencias motoras de manera representativa para el estudio clínico humano, en cambio su relevancia se basa en el estudio de la forma genética de la EP, enfocándose en genes específicos relacionados con la enfermedad23.
El gen α -syn codifica la proteína α -sinucleína, componente principal de los cuerpos de Lewy presentes en la EP. Ratones transgénicos presentan una disminución en los niveles de TH y dopamina, afectando su comportamiento14. En modelos con ratas se han dirigido factores virales con α -sinucleína, observando enfermedad en la proteína y neurodegeneración dopaminérgica, siendo un modelo ideal para probar nuevas estrategias neuroprotectoras36.
El gen LRRK2 es requerido para la supervivencia neuronal y su edición génica es la más común en los modelos de PD de tipo genético10. En los modelos con ratones se ha demostrado tener un poco o nulo daño en la función de las nDA en la SNPc14. En cambio, en ratones knock-out se ha logrado producir acumulación de α -sinucleína e inhibir la diferenciación de progenitores neuronales a nDA y aumentar la muerte celular10,37,38. En el caso de una sobreexpresión del gen se ha observado una ligera neurodegenereración de las nDA en la SNPc, pero sin alteración de los niveles de dopamina ni en la actividad locomotriz39.
Resultados de modelos en ratones sin el gen PINK1, gen esencial para la supervivencia neuronal bajo estrés oxidativo10, han demostrado una reducción gradual en los niveles de dopamina acompañada de una baja actividad locomotriz, sin la formación de cuerpos de Lewy o una degeneración nigroestriatal40. Por otra parte, los modelos utilizando Drosophila melanogaster con mutaciones en el gen PINK1 han mostrado defectos en la habilidad para volar y anormalidades en el complejo i mitocondrial41,42.
En el caso de los estudios realizados con ratones sin el gen de PARKIN, no se pudo comprobar anormalidades en el comportamiento de los animales, a pesar de la ligera disminución de la liberación de dopamina. Finalmente, modelos en ratones con mutaciones en el gen DJ-1, necesario para la resistencia contra estrés oxidativo, mostraron una disminución en la capacidad motora, una reducción de dopamina liberada en el estriado, pero no en la SNPc10,43 (tabla 1).
Modelos preclínicos de terapia celularActualmente los tratamientos más utilizados se basan en la sustitución de la dopamina o administración de agonistas de manera farmacológica, pero tienen la desventaja de perder su efectividad con el avance de la enfermedad y generan varios efectos secundarios45,46, además de la alternativa quirúrgica o estimulación profunda de cerebro, que a su vez puede generar hemorragia, infección y efectos secundarios neuropsiquiátricos47. La generación de nDAs a partir de CT para su trasplante representan un gran avance para el futuro de la terapia celular de enfermedades como la EP, ya que tiene como objetivos que las nDA injertadas puedan sobrevivir y formar conexiones con el cerebro de los pacientes, produciendo mejorías clínicas mesurables48,49. Sin embargo, para poder ser llevada a la clínica debe de ser probada en modelos preclínicos in vitro e in vivo y estandarizar factores críticos para la eficacia y seguridad del procedimiento, como la selección de pacientes, la colocación del injerto, su composición celular y regulación inmunológica48,50,51.
Algunas de las fuentes de tejido que se han utilizado para la obtención de nDA incluyen tejido mesencefálico ventral fetal humano (hfVM), médula adrenal, bulbo olfatorio, cuerpo carotídeo, células troncales embrionarias, células troncales neurales (NSC), células troncales mesenquimales, células troncales pluripotentes inducidas (iPSC) y células troncales partenogénicas humanas45,46.
Los trasplantes de médula adrenal fueron los primeros en ser estudiados para el tratamiento de la EP46. En un estudio clínico humano se realizaron trasplantes de médula adrenal, hfVM y tejido adrenal fetal. La médula adrenal generó mejorías de manera bilateral y simétricas, disminuyendo la rigidez, inestabilidad postural y alteraciones de marcha52,53. El hfVM generó mejorías considerables en la rigidez, inestabilidad postural, alteraciones de marcha, bradicinesia y expresión facial, aunque el temblor continuó manifestándose. El tejido adrenal fetal únicamente mostró disminución de rigidez y bradicinesia54. Sin embargo, dejaron de ser investigados debido a la alta mortalidad asociada a la cirugía abdominal y craneal46. Además del desarrollo de otros métodos terapéuticos, como el trasplante hfVMs, que pueden ser diferenciadas hacia nDAs, demostraron una alta recuperación histológica y funcional, sin formación de tumores en ratones y primates55. En estudios clínicos humanos en donde los pacientes recibieron trasplantes de manera bilateral en el putamen, se demostró que los injertos pueden sobrevivir a pesar del avance de la EP y el continuo tratamiento farmacológico, reinervando el estriado, restableciendo la liberación controlada de dopamina e integrándose en el circuito nigrostriatal48. A su vez, presenta complicaciones quirúrgicas en adición a la dificultad técnica de disección del tejido fetal resultando en una combinación de poblaciones celulares con alta mortalidad, sin mencionar la limitada disponibilidad de tejido fetal, complicaciones éticas y discinesias postoperatorias56,57.
Las NSC provenientes de adultos mantienen las propiedades de progenitores neurales característicos del sistema nervioso fetal. Sin embargo, su comportamiento es determinado por el ambiente extracelular en el área donde residen46,58. Al encontrarse en contacto con células endoteliales de vasos sanguíneos constituyen el nicho neurovascular, liberando factores que promueven su proliferación y neurogénesis59. En cambio, cercanas al bulbo olfatorio se encuentran células de tipo astrocíticas que tienen la capacidad de autorrenovarse y dar origen a progenitores de neuroblastos, aunque aún no se define su capacidad de formar nDAs funcionales46,55. Un estudio basado en que las células gliales envolventes olfatorias permiten una reentrada continua de las fibras axónicas en el bulbo olfatorio durante la adultez60, demostró que la combinación de trasplantes de nervios periféricos y centrales genera un andamio promotor de crecimiento axonal e innervación dopaminérgica en el estriado61. Otro tipo de NSC han sido descritas en el cuerpo carotídeo46, un órgano quimiosensorial derivado de la cresta neural compuesto de células glómicas tipo neuronales y envueltas en células de tipo-glial62. Las células tipo neuronales presentan altos niveles de dopamina almacenada en vesículas, del factor neurotrófico derivado del cerebro y GDNF, demostrando su contribución a la neurogénesis, neuroprotección y reemplazo de nDA63. Al ser trasplantados en la vía nigrostriatal de ratas parkinsonianas y primates no humanos tratados con MPTP muestran recuperación histológica y funcional, induciendo el brote de fibras nigrostriatales dopaminérgicas, sin efectos secundarios relacionados con el procedimiento quirúrgico62,64,65.
Las iPSC presentan la ventaja de generar nDA específicas de cada paciente sin conflictos éticos e inmunológicos, además de su alta diversidad de origen, aunque aún presentan probabilidades de mutagenicidad, daños en la integridad del genoma y formación de teratomas45,66,67.
Modelo de células troncales pluripotentes inducidasLas CT humanas son células no diferenciadas, que tienen la capacidad de autorrenovarse y diferenciarse a diferentes tipos celulares derivados de las 3 capas germinales: endodermo, mesodermo y ectodermo68. Algunos autores han clasificado las CT en 2 grupos: embrionarias y adultas o somáticas. Se diferencian en que presentan distintos grados de potencialidad, ya que pueden ser pluripotenciales, multipotenciales y/o células progenitoras de tejidos69. Las células pluripotenciales de tipo embrionario proceden del embrioblasto, que es la masa interna del blastocisto y se caracterizan por poder formar cualquier tipo de célula presente en un adulto, excepto los tejidos extraembrionarios70.
La nueva tecnología de las CT son las iPSC; estas células se derivan de células somáticas como los fibroblastos, que son reprogramadas con los factores de Yamanaka (Oct3/4, Sox2, c-Myc y Klf4) hacia un estado pluripotente71. Desde este nuevo estadio las iPSCs pueden ser diferenciadas hacia cualquier tipo celular presente en un adulto72. Estas células presentan un gran potencial para el modelado in vitro de enfermedades neurodegenerativas como la EP73.
Generación de neuronas dopaminérgicasLa generación de nDA se puede realizar a partir de células adultas reprogramadas de manera in vitro hacia un estado pluripotente. Posteriormente, estas células pueden ser diferenciadas hacia nDA, con la posibilidad de modificar genéticamente las iPSC para estudiar el efecto de un gen en específico, mediante el uso de protocolos de diferenciación neural establecidos que en su mayoría hacen uso de los factores y morfógenos expresados en el desarrollo normal de las nDA (Sonic Hedgehog, el factor de crecimiento de fibroblastos 8, el factor neurotrófico derivado del cerebro, entre otros). Las células obtenidas pueden ser utilizadas para investigación in vitro con el objetivo de realizar modelos celulares de la EP que mimeticen su patofisiología y sean útiles en el desarrollo de moléculas neuroprotectoras para el desarrollo de futuros tratamientos74–76. Una de las mayores ventajas de este modelo es que son compatibles con la mayoría de las técnicas de modificación genética disponibles actualmente: nucleasas de dedo de cinc (ZFN), las nucleasas efectoras tipo activador de la transcripción (TALEN) y el sistema de repetición palindrómica corta entrecruzada regularmente (CRISPR)/CRISPR-Cas9 (fig. 1)77.
 para la enfermedad de Parkinson (EP). Los modelos de la EP utilizando la tecnología de iPSC comienzan con la obtención de células somáticas de pacientes con la EP. Una vez que se establecen los cultivos de células somáticas, p. ej. fibroblastos, se procede a realizar la reprogramación celular para obtener iPSCs utilizando vectores, p. ej. Sendai, lentivirus, retrovirus y los factores de transcripción, Oct3/4, Sox2, c-Myc y Klf-4. Consecuentemente a la reprogramación celular exitosa, se dirige la diferenciación hacia el destino neuronal dopaminérgico adicionando factores de transcripción, p. ej. Sonic Hedgehog, factor de crecimiento de fibroblastos 8, factor neurotrófico derivado del cerebro. Adicionalmente, en el estado pluripontente las iPSC pueden estar sujetas a modificaciones genéticas para sobreexpresar o inhibir genes de interés clínico. Finalmente, las neuronas dopaminérgicas generadas son utilizadas como modelo de la EP y realizar estudios in vivo, in vitro o criopreservación para su futuro uso, dirigiendo al descubrimiento de futuros tratamientos.")
Diagrama descriptivo de generación de modelos de células troncales pluripotentes inducidas (iPSC) para la enfermedad de Parkinson (EP).
Los modelos de la EP utilizando la tecnología de iPSC comienzan con la obtención de células somáticas de pacientes con la EP. Una vez que se establecen los cultivos de células somáticas, p. ej. fibroblastos, se procede a realizar la reprogramación celular para obtener iPSCs utilizando vectores, p. ej. Sendai, lentivirus, retrovirus y los factores de transcripción, Oct3/4, Sox2, c-Myc y Klf-4. Consecuentemente a la reprogramación celular exitosa, se dirige la diferenciación hacia el destino neuronal dopaminérgico adicionando factores de transcripción, p. ej. Sonic Hedgehog, factor de crecimiento de fibroblastos 8, factor neurotrófico derivado del cerebro. Adicionalmente, en el estado pluripontente las iPSC pueden estar sujetas a modificaciones genéticas para sobreexpresar o inhibir genes de interés clínico. Finalmente, las neuronas dopaminérgicas generadas son utilizadas como modelo de la EP y realizar estudios in vivo, in vitro o criopreservación para su futuro uso, dirigiendo al descubrimiento de futuros tratamientos.
A partir del siglo pasado se comenzaron a realizar experimentos para comprender y tratar la EP, utilizado diferentes enfoques para su análisis y generando información para el avance científico. Con el descubrimiento de Yamanaka se ha explotado la investigación sobre sus aplicaciones en la EP72,78,79. El desarrollo de modelos preclínicos que utilizan iPSC ha aumentado de manera considerable en la última década, y cada uno de ellos nos acerca cada vez más a entender mejor la fisiopatología de la EP72,75. La investigación sobre el desarrollo de la EP de tipo familiar se ha basado en el estudio de factores genéticos involucrados en las patogénesis del trastorno (tabla 2)79. Principalmente, se busca inducir una disminución de las funciones motoras características de la enfermedad de Parkinson, como son la bradiquinesia, los temblores, la rigidez y el comportamiento, así como el papel de algunos genes que han demostrado estar involucrados en su desarrollo, incluyendo: α -SNCA, LRKK2, PINK1, PARKIN y GBA114,35,81,82,83–86.
Modelos genéticos de iPSC de la enfermedad de Parkinson. Efectos generados por alteraciones de genes involucrados en la patogénesis de la enfermedad de Parkinson en modelos utilizando iPSC
| Gen alterado | Efecto | Ref. |
|---|---|---|
| SNCA | Acumulación o sobreexpresión de la proteína α -sinucleína en las nDA | Byers et al.84 |
| LRRK2 | Reducción del crecimiento de las neuritasAumento del estrés oxidativoDeterioro de la capacidad de autorrenovación y diferenciación neuronal de iPSCs | Shi et al.85 |
| PINK1/PARKIN | Fenotipos anormales de mitofagia y autofagiaMayor vulnerabilidad al estrés | Shaltouki et al.86,, Pickrell y Youle87 |
| GBA1 | Altos niveles de α -sinucleína, autofagia, defectos lisosomales, desbalance en la homeostasis del calcio, reducción de la toma y almacenamiento de dopamina | Schöndorf et al.83 |
El gen de SNCA se ha relacionado con la acumulación o sobreexpresión de la proteína α -sinucleína en las nDA84. En cuanto al gen LRRK2 se ha descubierto que al perder su funcionalidad se reduce el crecimiento de las neuritas, aumenta el estrés oxidativo, hay un daño de ADN, además de deteriorar la capacidad de autorrenovación y diferenciación neuronal de iPSC85. Se ha demostrado una correlación entre los genes de PINK1 y Parkina debido a que ambos contribuyen para la homeostasis celular y mitocondrial en nDA. Adicionalmente, nDA derivadas de iPSC que presentan mutaciones en los genes PINK1 o Parkina muestran fenotipos anormales de mitofagia y autofagia, además de mayor vulnerabilidad al estrés86,87. Finalmente, las mutaciones del gen GBA1 han indicado una alta relación con altos niveles de α -sinucleína, autofagia, defectos lisosomales, desbalance en la homeostasis del calcio, reducción de la toma y almacenamiento de dopamina en nDA derivadas de iPSC83. En adición, las alteraciones epigenéticas afectan la metilación del ADN en los pacientes induciendo un recambio erróneo de proteínas y variaciones en la morfología celular88.
En la actualidad se han obtenido líneas celulares de iPSC derivadas de pacientes con la EP, brindando la ventaja de que los fenotipos específicos de la EP son originados por la genética de los pacientes desde las primeras etapas de la enfermedad, ya sea en casos de EP hereditarios o esporádicos74,89. La mayoría de los casos de la EP son de tipo esporádico, del cual ha sido más complicado definir su etiología, ya que no se conocen mutaciones genéticas específicas relacionadas con esta forma80, por lo que su investigación se ha centrado en la diferenciación de nDAs derivadas de iPSCs de pacientes con EP esporádica90.
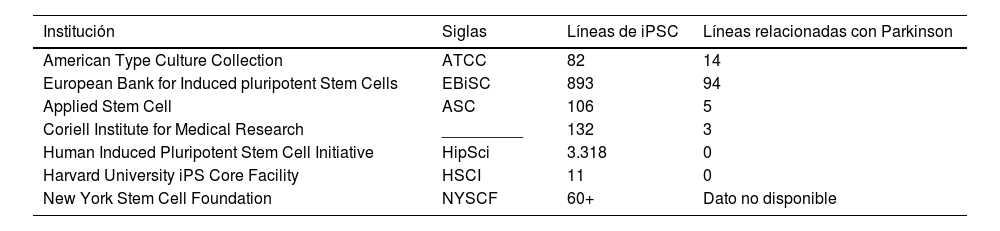
Líneas celulares relacionadas con la EP pueden encontrarse en diversos bancos celulares (tabla 3). Se han realizado varios trabajos de investigación utilizando estos métodos, donde se creó una biblioteca con más de 60 líneas iPSC utilizando el método de reprogramación celular con 3 vectores diferentes: lentivirus, retrovirus y virus Sendai91,92.
Cuadro demostrativo de algunas instituciones de bancos celulares. Existen varias instituciones que brindan la función de almacenamiento de líneas celulares alrededor del mundo, algunas de las más conocidas se encuentran descritas en el siguiente cuadro, señalando la cantidad de iPSC disponibles y aquellas relacionadas con la enfermedad de Parkinson
| Institución | Siglas | Líneas de iPSC | Líneas relacionadas con Parkinson |
|---|---|---|---|
| American Type Culture Collection | ATCC | 82 | 14 |
| European Bank for Induced pluripotent Stem Cells | EBiSC | 893 | 94 |
| Applied Stem Cell | ASC | 106 | 5 |
| Coriell Institute for Medical Research | _________ | 132 | 3 |
| Human Induced Pluripotent Stem Cell Initiative | HipSci | 3.318 | 0 |
| Harvard University iPS Core Facility | HSCI | 11 | 0 |
| New York Stem Cell Foundation | NYSCF | 60+ | Dato no disponible |
Existen varias instituciones que brindan la función de almacenamiento de líneas celulares alrededor del mundo, algunas de las más conocidas se encuentran descritas en esta tabla, señalando la cantidad de iPSC disponibles y aquellas relacionadas con la enfermedad de Parkinson.
Estudios realizados con iPSC en modelos murinos han demostrado la misma potencia y eficacia que las nDA obtenidas de tejido fetal, ya que muestran una alta capacidad de crecimiento axonal específico a larga distancia, una diferenciación rápida, eficiente y sincronizada, evitando la formación de tumores93. Recientemente, se ha demostrado que estas células apoyan la recuperación funcional de los déficits inducidos por lesiones, posicionando a las iPSC como el futuro de la investigación de una amplia gama de enfermedades8.
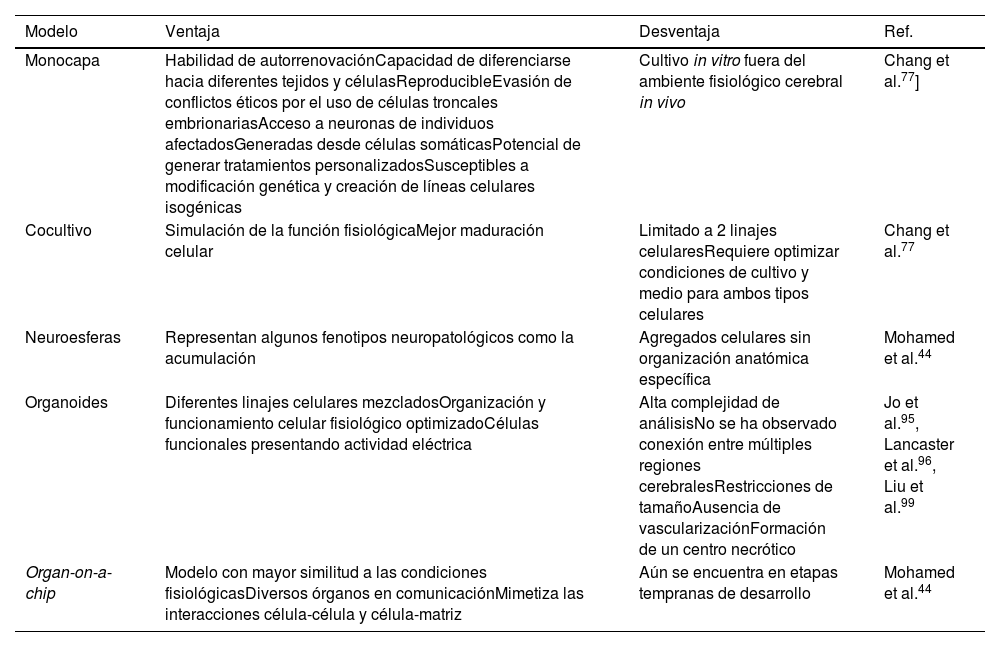
Los modelos celulares de crecimiento en monocapa fueron los primeros en ser desarrollados, con el objetivo de estudiar los mecanismos celulares y moleculares individualmente52. Sin embargo, pueden no ser completamente representativos de la complejidad de las enfermedades neurodegenerativas, por lo que se ha recurrido a métodos de cocultivo y modelos 3D (tabla 4)77. El método de cocultivo ha generado buenos avances al generar un ambiente en el que 2 tipos celulares, como astrocitos y neuronas, mimetizan de manera similar al fisiológico la actividad celular, al presentarse interacciones intercelulares y una matriz extracelular mixta52. Ya que los astrocitos brindan soporte estructural y metabólico a las neuronas80, se ha observado un aumento en el ritmo de maduración de las neuronas, mayor cantidad de marcadores neurales y estabilización de la función mitocondrial al disminuir la producción de especies reactivas de oxígeno94. La desventaja de este método es la limitación de los linajes celulares a utilizar y la necesidad de optimizar las condiciones de cultivo funcionales para ambas77.
Modelos de la enfermedad de Parkinson. Comparación de diferentes modelos con iPSCs de la enfermedad de Parkinson con sus respectivas ventajas y desventajas
| Modelo | Ventaja | Desventaja | Ref. |
|---|---|---|---|
| Monocapa | Habilidad de autorrenovaciónCapacidad de diferenciarse hacia diferentes tejidos y célulasReproducibleEvasión de conflictos éticos por el uso de células troncales embrionariasAcceso a neuronas de individuos afectadosGeneradas desde células somáticasPotencial de generar tratamientos personalizadosSusceptibles a modificación genética y creación de líneas celulares isogénicas | Cultivo in vitro fuera del ambiente fisiológico cerebral in vivo | Chang et al.77] |
| Cocultivo | Simulación de la función fisiológicaMejor maduración celular | Limitado a 2 linajes celularesRequiere optimizar condiciones de cultivo y medio para ambos tipos celulares | Chang et al.77 |
| Neuroesferas | Representan algunos fenotipos neuropatológicos como la acumulación | Agregados celulares sin organización anatómica específica | Mohamed et al.44 |
| Organoides | Diferentes linajes celulares mezcladosOrganización y funcionamiento celular fisiológico optimizadoCélulas funcionales presentando actividad eléctrica | Alta complejidad de análisisNo se ha observado conexión entre múltiples regiones cerebralesRestricciones de tamañoAusencia de vascularizaciónFormación de un centro necrótico | Jo et al.95, Lancaster et al.96, Liu et al.99 |
| Organ-on-a-chip | Modelo con mayor similitud a las condiciones fisiológicasDiversos órganos en comunicaciónMimetiza las interacciones célula-célula y célula-matriz | Aún se encuentra en etapas tempranas de desarrollo | Mohamed et al.44 |
Fuente: Mohamed et al.44
Una de las nuevas tendencias de investigación es la generación de organoides 3D y neuroesferas utilizando iPSC, ya que presentan las ventajas de que las células iPSC se diferencian de manera más espontánea, en un periodo de uno a 2 meses, en tipos neuronales funcionales, mimetizando de mejor manera el desarrollo del cerebro, así como las neuropatologías95,96. Los organoides pueden conservarse en cultivo hasta por un año, a pesar de que a los 6 meses comienzan a encogerse97. Dentro de las limitaciones que presentan se incluyen las restricciones de tamaño, la ausencia de vascularización, su corta duración, la formación de un centro necrótico, la imprecisión en la identificación de las regiones cerebrales, la variabilidad por lote y su dificultad técnica98. Estos modelos también son utilizados para estudiar los mecanismos fisiológicos de la EP, posibles tratamientos y el desarrollo de la medicina personalizada99. Finalmente tenemos los modelos de organ-on-a-chip utilizando iPSC, que consisten en un sistema microfisiológico in vitro donde varios organoides son cultivados de manera que a través de un flujo líquido se genere un contacto intercelular para mimetizar las condiciones fisiológicas del cuerpo, incluyendo interacciones célula-célula y célula-matriz; este modelo se encuentra en etapas tempranas de desarrollo y representa un descubrimiento prometedor44.
Ventajas de las células troncales pluripotentes inducidas sobre otros modelosLas CT pueden ser destinadas a diferenciarse hacia nDA de la SNPc para modelar la EP a nivel celular. Sin embargo, las iPSCs presentan la ventaja de que las células son específicas de los pacientes, evitando así la necesidad de utilizar inmunosupresores al realizar trasplantes y el sacrificio humano para la obtención de células troncales embrionarias, además de la posibilidad de corregir genéticamente las células para producir fenotipos funcionales100. El proceso de diferenciación de iPSC a nDA mimetiza el desarrollo embrionario de estas células, se conserva la maquinaria celular endógena y los mecanismos de transcripción101. Adicionalmente se evita el uso de neurotoxinas, manteniendo un desarrollo natural de la enfermedad sin estímulos estresores externos10.
Dentro de las áreas a mejorar en la investigación en edición genética está la diferenciación óptima de nDA, que depende de reguladores moleculares de la función y estabilidad de las proteínas involucradas en la EP102. Las técnicas más utilizadas para realizar modificaciones genéticas todavía presentan varias limitaciones, dentro de las cuales se incluyen: 1) la unión de extremos no homólogos, propensa a errores que pueden resultar en la inestabilidad del genoma y mutaciones causantes de enfermedades103; 2) la recombinación dirigida por homología utilizando ZFN o TALEN, presenta problemas de capacidad de focalización debido a la disponibilidad de bibliotecas para ZFN, además de la complejidad y el tamaño de los vectores requeridos para TALEN104; y 3) CRISPR-Cas9 ha tenido una gran explosión de avances, pero al ser una técnica nueva aún requiere estudiar las enzimas Cas9 para su correcta dirección105. La mejora de las técnicas de edición de genes en tejidos específicos y poblaciones de células in vivo sigue siendo uno de los principales retos para la terapia génica humana segura y eficaz106.
Una de las dificultades de la terapia celular a través de trasplantes es la formación de cuerpos de Lewy con el paso de tiempo, con la reaparición de los síntomas motores. Esta desventaja puede ser superada utilizando la nueva tecnología de CRISPR/Cas9, con la que se desarrollaron nDAs derivadas de iPSCs a las que se les realizó una deleción en el gen SNCA, que al estar en contacto con fibrillas preformadas de α -sinucleína presentan resistencia a la formación de agregados de esta proteína de manera permanente50.
En 2018 se inició la primera terapia para la EP trasplantando precursores de nDA derivadas de iPSC en la SNPc del cerebro. Para este procedimiento se seleccionaron 7 pacientes con un nivel de Parkinson moderado, el primer paciente no ha mostrado efectos adversos y, de continuar así, se espera proseguir con el resto de los pacientes el presente año107,108. Antes de proseguir con los estudios clínicos en pacientes humanos realizaron pruebas en ratones para analizar la tumorigenicidad, toxicidad, biodistribución de las nDA obtenidas y formación de teratomas, adicionalmente se observó en ratas lesionadas con la neurotoxina 6-OHDA la disminución de movimientos asimétricos rotacionales hasta niveles normales y finalmente comprobaron la supervivencia de los trasplantes neuronales, sin efectos secundarios en primates no humanos109. Este proyecto representa un gran paso en el desarrollo de tratamientos de la EP, que en caso de ser exitoso abre las puertas a un nuevo futuro como tratamiento.
Actualmente se encuentran 2 estudios clínicos en progreso registrados en el instituto nacional de salud (NIH) para investigar la aplicación de iPSC en pacientes con la EP. El Centro Clínico de los Institutos Nacionales de Salud (CC) (Instituto Nacional del Corazón, Pulmones y Sangre), identificado en clinicaltrials.gov con el registro NCT01143454, inició un proyecto de pruebas clínicas desde junio de 2010 que continúa hasta la fecha, nombrado «Caracterización de pacientes con presentaciones infrecuentes y/o enfermedades poco frecuentes asociadas al sistema cardiovascular». El objetivo consiste en caracterizar la etiología molecular, fisiopatología y la historia de enfermedades poco comunes conocidas y desconocidas, entre ellas la EP, que se presentan con síntomas y signos asociados al riesgo de disfunción cardiovascular manifiesta o potencial, haciendo uso de material biológico y recolecta de tejidos, para perfeccionar los protocolos de diagnóstico110.
El segundo estudio titulado «Desarrollo de iPS a partir de células somáticas donadas de pacientes con enfermedades neurológicas», realizado desde abril de 2009 hasta la fecha por la organización médica Hadassah, identificado en crinicaltrials.gov bajo el registro NCT00874783, tiene como propósito desarrollar células iPS humanas a partir de cultivos celulares de biopsias de piel o del cabello de pacientes a través de la expresión forzada de factores de transcripción. Principalmente serán empleadas para modelar enfermedades neurodegenerativas como la EP, probar fármacos, generar información importante para la investigación básica y el desarrollo de tecnología que eventualmente permita el uso de células iPS para futuras terapias de trasplantes111.
Los modelos preclínicos basados en iPSC presentan algunas limitaciones, como el mejoramiento de la estandarización de los protocolos de obtención, reprogramación, diferenciación, seguridad y aplicación a pacientes, el potencial de formación de tumores y teratomas, la necesidad de desarrollar métodos de inducción con tecnología no integrativa a ADN más eficientes y rápidos112. A pesar de estas limitaciones, los modelos con iPSCs actualmente representan la mayor similitud a la EP fenotípicamente, permite estudiar los efectos celulares de mutaciones en tiempo real, cuantificar efectos del estrés oxidativo celular y mitocondrial y analizar fármacos o moléculas con potencial neuroprotector10,113–116.
Discusión o perspectivasLa EP es un trastorno de interés mundial del cual no se conoce exactamente su etiología, al ser principalmente multifactorial en el tipo esporádico, ni se cuenta con terapias efectivas para el tratamiento de la enfermedad. A pesar de que los medicamentos disponibles aumentan la producción de dopamina en las neuronas dopaminérgicas en la SNPc del cerebro, solo disminuyen los síntomas de la enfermedad, pero no reducen o previenen la progresión de la EP.
Las iPSC pueden proporcionar un modelo fundamental para la investigación de enfermedades neurodegenerativas como el Parkinson y probar posibles fármacos para su tratamiento. Las neuronas dopaminérgicas derivadas de iPSC son capaces de reproducir el desarrollo de la EP con el fin de estudiar la progresión de la enfermedad y detectar posibles marcadores moleculares que funcionen como indicadores para su oportuno diagnóstico. Además de brindar una amplia gama de opciones para desarrollar los modelos, ya sea desde un enfoque toxicológico o genético, in vitro, in vivo o cocultivo y, en caso de ser necesario, varias especies animales disponibles.
Es necesario resaltar que aún es necesario perfeccionar los estándares requeridos para el uso de iPSCs en trasplantes como tratamiento de la EP, ya sea para fines de investigación o aplicaciones clínicas. Uno de los grandes retos a los que se enfrentan los modelos en iPSCs es que la enfermedad de Parkinson se presenta principalmente en la población de la tercera edad, y al reprogramar las células somáticas se genera un estado similar al embrionario, por lo que se deben reproducir estas características de la senescencia neuronal en el modelo in vitro.
Sin embargo, con las herramientas de modificaciones epigenéticas, de cromatina y regulación genómica de genes relevantes para la EP, los modelos de iPSC avanzan hacia la medicina personalizada. Líneas celulares de pacientes pueden ser generadas a partir de células somáticas, evadiendo cuestiones éticas relacionadas con el uso de células troncales embrionarias, además de la posibilidad de probar medicamentos de forma específica a pacientes de manera in vitro.
FinanciaciónEste trabajo fue apoyado por fondos del Consejo Nacional de Ciencia y Tecnología (CONACyT; No 300638, 271307 y FODECIJAL; No. 8084-2019).
Conflicto de interesesNinguno.

