








Las ataxias espinocerebelosas de herencia recesiva constituyen un amplio grupo de enfermedades del cerebelo y/o de sus conexiones; en muchos casos también se afectan otras partes del sistema nervioso. Asimismo, y con cierta frecuencia, se acompañan de diversas manifestaciones sistémicas (cardiopatía, alteraciones cutáneas, endocrinopatías, malformaciones esqueléticas).
DesarrolloEn este trabajo se revisan los conocimientos actuales sobre las principales ataxias recesivas de curso progresivo, con el fin de establecer unas claves que faciliten su complejo diagnóstico.
ConclusionesUna cuidadosa evaluación clínica (síndrome espinocerebeloso puro o plus con o sin manifestaciones sistémicas), acompañada de la determinación de ciertos marcadores de laboratorio (vitamina E, acantocitosis, alfa-fetoproteína, ácido láctico, albúmina, colesterol, ácido fitánico, coenzima-Q10, CK, colestanol, quitotriosidasa, cobre, ceruloplasmina), la valoración de los datos del estudio electroneuromiográfico (ausencia o presencia de neuropatía y tipo de la misma) y los hallazgos del estudio de resonancia magnética (ausencia o presencia de atrofia cerebelosa, ausencia o presencia de alteraciones de la intensidad de señal y localizaciones de las mismas), ayudarán al clínico a establecer unas determinadas sospechas diagnósticas, que siempre procurará confirmar con la detección de la mutación genética causal. El hallazgo de la mutación es decisivo para establecer el pronóstico y consejo genético, además permitirá indicar un tratamiento eficaz en determinadas entidades (abetalipoproteinemia, ataxia por déficit de vitamina E, enfermedad de Refsum, xantomatosis cerebrotendinosa, enfermedad de Niemann-Pick tipo C, enfermedad de Wilson). Sin diagnóstico genético no será posible realizar investigación básica ni tampoco poner en marcha ensayos terapéuticos.
Autosomal recessive spinocerebellar ataxia refers to a large group of diseases affecting the cerebellum and/or its connections, although they may also involve other regions of the nervous system. These diseases are accompanied by a wide range of systemic manifestations (cardiopathies, endocrinopathies, skeletal deformities, and skin abnormalities).
DevelopmentThis study reviews current knowledge of the most common forms of autosomal recessive spinocerebellar ataxia in order to provide tips that may facilitate diagnosis.
ConclusionsA thorough assessment of clinical phenotype (pure cerebellar or cerebellar-plus syndrome, with or without systemic manifestations), laboratory tests (vitamin E, acanthocytosis, albumin, cholesterol, phytanic acid, lactic acid, creatine kinase, cholestanol, coenzyme Q10, alpha-fetoprotein, copper, ceruloplasmin, chitotriosidase), nerve conduction studies (presence and type of neuropathy), and an magnetic resonance imaging study (presence of cerebellar atrophy, presence and location of signal alterations) may help establish a suspected diagnosis, which should be confirmed by detecting the underlying genetic mutation. A positive genetic test result is necessary to determine prognosis and provide adequate genetic counselling, and will also permit appropriate treatment of some entities (abetalipoproteinaemia, ataxia with vitamin E deficiency, Refsum disease, cerebrotendinous xanthomatosis, Niemann-Pick disease type C, Wilson disease). Without a genetic diagnosis, conducting basic research and therapeutic trials will not be possible.
Trastorno del equilibrio y falta de coordinación motora son 2 acepciones principales de la palabra ataxia (en griego significa falta de orden). Este término fue empleado por vez primera, en 1864, por Duchenne de Boulogne1, quien denominó «ataxia locomotriz» al trastorno de la marcha producido por la tabes dorsalis. No obstante, ya un año antes Nikolaus Friedreich2 había descrito la enfermedad que lleva su nombre, y que constituye la variedad más frecuente de heredoataxia recesiva. A finales del siglo xix, Pierre Marie separó la ataxia de Friedreich (AF) de las ataxias de herencia dominante.
Las ataxias degenerativas producen una alteración progresiva de la función motora, derivada de la afectación del cerebelo y/o de sus conexiones principales (corteza cerebral, cordones posteriores y haces espinocerebelosos, núcleos vestibulares, núcleo rojo, núcleos pontinos y oliva bulbar inferior)3. Además de los síntomas de desequilibrio (ataxia de línea media), descoordinación de extremidades (dismetría/disdiacocinesia), disartria, oscilopsia y nistagmo, los pacientes con ataxia pueden presentar retraso mental y deterioro cognitivo, epilepsia, piramidalismo, movimientos anormales, alteraciones visuales, hipoacusia, enfermedad de motoneurona inferior, neuropatía periférica y miopatía. El cerebelo está también implicado en funciones ejecutivas, procesado de las emociones y del lenguaje, que pueden alterarse en las heredoataxias4–6.
En este trabajo, basado en una búsqueda bibliográfica realizada en PUBMED, OMIN, Gene-Reviews, y con la aportación de datos e iconografía de casos personales, pretendemos establecer una sistematización en el diagnóstico de las ataxias degenerativas progresivas de herencia recesiva (se excluyen las ataxias congénitas como los síndromes de Joubert, Norman, Gillespie, Pollit, etc.), con especial énfasis en las más frecuentes y en aquellas en la que se dispone de terapéutica eficaz.
DesarrolloEl paciente que consulta por ataxiaMediante la historia clínica se perfilará el comienzo de la ataxia, el tiempo y perfil evolutivo (estática, progresiva o intermitente), el tipo de síndrome espinocerebeloso (de línea media, apendicular o mixto) y la existencia o no de afectación de otros sistemas (síndrome cerebeloso puro o plus), además de los antecedentes personales (tóxicos, fármacos, enfermedades carenciales, endocrinopatías, infecciones, neoplasias, padecimientos autoinmunes, prionopatías)7, y la historia familiar con el tipo de herencia que muestra (dominante, recesiva, ligada a X, mitocondrial matrilineal) si fuese positiva. El acrónimo SCA (Spino Cerebellar Ataxia) se utiliza para denominar a las heredoataxias progresivas de herencia dominante. Por otra parte, ARCA (Autosomal Recessive Ataxia) designa las ataxias de herencia recesiva en las que predomina la atrofia cerebelosa. Las escalas clínicas scale for the assessment and rating of ataxia (SARA)8 e international cooperative ataxia rating scale (ICARS)9 son útiles para el control evolutivo, y las escalas de calidad de vida, como la EQ-5D, para valorar las repercusiones de la enfermedad en la salud.
Los fallos o retrasos en el diagnóstico de las ataxias se producen por diversas causas y situaciones: el enfermo niega o minimiza sus síntomas; el desequilibrio es achacado a trastorno emocional o a estrés; los pacientes son remitidos a otros especialistas como traumatólogos, reumatólogos, oftalmólogos y otorrrinolaringólogos; tras una consulta inicial de un caso incipiente no se producen nuevas reevaluaciones10.
En un paciente que consulta por ataxia, el número de estudios a realizar para llegar al diagnóstico puede ser muy amplio y dependerá de la hipótesis diagnóstica planteada después de realizada la historia clínica. En la tabla 1 se detalla toda la batería de técnicas complementarias, que deberán utilizarse selectivamente en cada caso.
Pruebas que puede ser necesario realizar en un paciente con ataxia progresiva de origen no aclarado
| Neuroimagen |
| RM cerebral y medular |
| TAC toracoabdominal |
| PET |
| DAT-scan |
| Tomografía de coherencia óptica |
| Estudios neurofisiológicos |
| Electromiograma |
| Potenciales evocados visuales, auditivos, somatosensoriales y motores |
| Reflejo del parpadeo |
| Audiometría y electronistagmografía |
| Electroencefalograma |
| Estudios de laboratorio |
| A) Sangre/suero/orina: hemograma, frotis (acantocitos), VSG, bioquímica, proteinograma, lipidograma, lactato, piruvato, alfa-fetoproteína, ECA, CK, TSH, ácido fitánico, colestanol, quitotriosidasa, vitamina E, vitamina B12, ácido fólico, TSH, amonio, ceruloplasmina, cobre, plomo, cinc. Panel inmunológico (incluir antitiroideos, antigliadina). Panel de síndrome paraneoplásico. Estudios microbiológicos (lúes, Lyme, virus hepatotropos, VIH, HTLV). Ácidos grasos de cadena larga. Co-Q10. Aminoácidos y ácidos orgánicos. Hidrolasas lisosomales (hexosaminidasa incluida) |
| B) Estudio de LCR: células, proteínas, glucosa, IgG/bandas oligoclonales, antígeno criptocócico, cultivo, PCR para diversos microorganismos |
| Estudios genéticos |
| Biopsias (nervio, piel, conjuntiva, intestinal, médula ósea) |
CK: creatincinasa; Co-Q10: coenzima Q10; ECA: enzima convertidora de la angiotensina; HTLV: virus linfotrópico de células T humanas; IgG: inmunoglobulina G; LCR: líquido cefalorraquídeo; PCR: proteína C reactiva; PET: tomografía por emisión de positrones; RM: resonancia magnética; TAC: tomografía axial computarizada; TSH: hormona estimulante de la tiroides; VIH: virus de la inmunodeficiencia humana; VSG: velocidad de sedimentación glomerular.
Las ataxias recesivas suelen comenzar antes de los 30 años, aunque hay casos de inicio neonatal y otros tardíos; a menudo, su progresión es más rápida que la de las dominantes. Su cuadro clínico puede ser un síndrome cerebeloso puro, pero lo más habitual es que, al lado de la clínica de disfunción cerebelosa, el paciente presente manifestaciones derivadas de la afectación de otros sistemas tales como retraso mental, deterioro cognitivo, epilepsia, trastornos del movimiento (distonía, corea, parkinsonismo, temblor), piramidalismo, trastornos oculomotores (apraxia oculomotora, parálisis supranuclear de la mirada vertical), neuropatía periférica, afectación retiniana e hipoacusia. También es importante considerar la afectación de otros órganos (miocardiopatía, diabetes, hepatoesplenomegalia, afectación músculo-esquelética)10–15.
La AF es la más frecuente de las ataxias recesivas, seguida de la ataxia telangiectasia (AT), la ataxia con apraxia oculomotora tipo i (AOA1) y la ataxia espástica de Charlevoix-Saguenay (ARSACS)15,16. Algunos tipos de ataxias recesivas tienen marcadores bioquímicos. Los estudios de neuroimagen, fundamentalmente la resonancia magnética (RM), pueden aportar datos claves para el diagnóstico de algunas ataxias recesivas, tal como se expondrá en el apartado de descripción particular. El estudio electrofisiológico de los nervios periféricos es muy útil para descartar o confirmar la presencia de neuropatía y el tipo de la misma (motora, sensitiva, axonal, desmielinizante, mixta).
En un grupo de ataxias recesivas causadas por defectos metabólicos (abetalipoproteinemia [ABLP], ataxia por déficit de vitamina E [ADVE], enfermedad de Refsum, xantomatosis cerebrotendinosa [XC], enfermedad de Niemann-Pick tipo C, enfermedad de Wilson, y ARCAII) se dispone de tratamientos con capacidad de modificar su curso17; este tipo de ataxias deben siempre tenerse en cuenta a la hora del diagnóstico diferencial10. Otros trastornos metabólicos, como la deficiencia de biotinidasa, los trastornos del ciclo de la urea, la enfermedad de Hartnup, la deficiencia de alfa-metil-CoA-racemasa y la deficiencia de gamma-glutamil-cisteína-sintetasa también pueden producir cuadros multisistémicos, entre los que se incluyen ataxias tempranas.
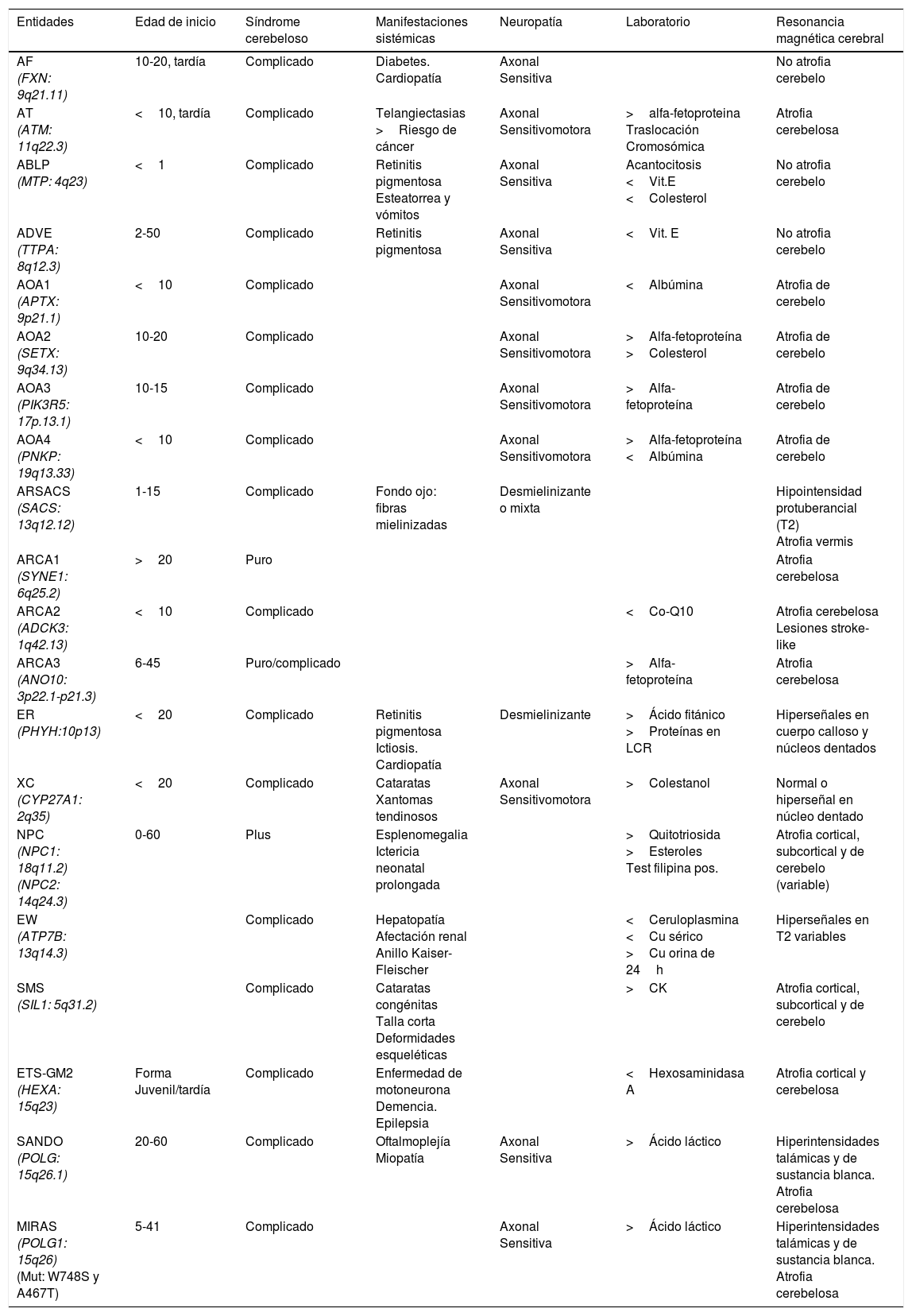
En la tabla 2 se resumen los datos clínicos, electromiográficos, de laboratorio y RM, de las principales heredoataxias recesivas.
Principales entidades de herencia recesiva y genética conocida que pueden dar lugar a un cuadro de ataxia progresiva
| Entidades | Edad de inicio | Síndrome cerebeloso | Manifestaciones sistémicas | Neuropatía | Laboratorio | Resonancia magnética cerebral |
|---|---|---|---|---|---|---|
| AF (FXN: 9q21.11) | 10-20, tardía | Complicado | Diabetes. Cardiopatía | Axonal Sensitiva | No atrofia cerebelo | |
| AT (ATM: 11q22.3) | <10, tardía | Complicado | Telangiectasias >Riesgo de cáncer | Axonal Sensitivomotora | >alfa-fetoproteina Traslocación Cromosómica | Atrofia cerebelosa |
| ABLP (MTP: 4q23) | <1 | Complicado | Retinitis pigmentosa Esteatorrea y vómitos | Axonal Sensitiva | Acantocitosis <Vit.E <Colesterol | No atrofia cerebelo |
| ADVE (TTPA: 8q12.3) | 2-50 | Complicado | Retinitis pigmentosa | Axonal Sensitiva | <Vit. E | No atrofia cerebelo |
| AOA1 (APTX: 9p21.1) | <10 | Complicado | Axonal Sensitivomotora | <Albúmina | Atrofia de cerebelo | |
| AOA2 (SETX: 9q34.13) | 10-20 | Complicado | Axonal Sensitivomotora | >Alfa-fetoproteína >Colesterol | Atrofia de cerebelo | |
| AOA3 (PIK3R5: 17p.13.1) | 10-15 | Complicado | Axonal Sensitivomotora | >Alfa-fetoproteína | Atrofia de cerebelo | |
| AOA4 (PNKP: 19q13.33) | <10 | Complicado | Axonal Sensitivomotora | >Alfa-fetoproteína <Albúmina | Atrofia de cerebelo | |
| ARSACS (SACS: 13q12.12) | 1-15 | Complicado | Fondo ojo: fibras mielinizadas | Desmielinizante o mixta | Hipointensidad protuberancial (T2) Atrofia vermis | |
| ARCA1 (SYNE1: 6q25.2) | >20 | Puro | Atrofia cerebelosa | |||
| ARCA2 (ADCK3: 1q42.13) | <10 | Complicado | <Co-Q10 | Atrofia cerebelosa Lesiones stroke-like | ||
| ARCA3 (ANO10: 3p22.1-p21.3) | 6-45 | Puro/complicado | >Alfa-fetoproteína | Atrofia cerebelosa | ||
| ER (PHYH:10p13) | <20 | Complicado | Retinitis pigmentosa Ictiosis. Cardiopatía | Desmielinizante | >Ácido fitánico >Proteínas en LCR | Hiperseñales en cuerpo calloso y núcleos dentados |
| XC (CYP27A1: 2q35) | <20 | Complicado | Cataratas Xantomas tendinosos | Axonal Sensitivomotora | >Colestanol | Normal o hiperseñal en núcleo dentado |
| NPC (NPC1: 18q11.2) (NPC2: 14q24.3) | 0-60 | Plus | Esplenomegalia Ictericia neonatal prolongada | >Quitotriosida >Esteroles Test filipina pos. | Atrofia cortical, subcortical y de cerebelo (variable) | |
| EW (ATP7B: 13q14.3) | Complicado | Hepatopatía Afectación renal Anillo Kaiser-Fleischer | <Ceruloplasmina <Cu sérico >Cu orina de 24h | Hiperseñales en T2 variables | ||
| SMS (SIL1: 5q31.2) | Complicado | Cataratas congénitas Talla corta Deformidades esqueléticas | >CK | Atrofia cortical, subcortical y de cerebelo | ||
| ETS-GM2 (HEXA: 15q23) | Forma Juvenil/tardía | Complicado | Enfermedad de motoneurona Demencia. Epilepsia | <Hexosaminidasa A | Atrofia cortical y cerebelosa | |
| SANDO (POLG: 15q26.1) | 20-60 | Complicado | Oftalmoplejía Miopatía | Axonal Sensitiva | >Ácido láctico | Hiperintensidades talámicas y de sustancia blanca. Atrofia cerebelosa |
| MIRAS (POLG1: 15q26) (Mut: W748S y A467T) | 5-41 | Complicado | Axonal Sensitiva | >Ácido láctico | Hiperintensidades talámicas y de sustancia blanca. Atrofia cerebelosa |
ABLP: abetalipoproteinemia; ADVE: ataxia por déficit de vitamina E; AF: ataxia de Friedreich; ARCA: ataxia recesiva cerebelosa; AT: ataxia telangiectasia; AOA: ataxia con apraxia oculomotora; ARSACS: ataxia recesiva de Charlevoix-Saguenay; CK: creatincinasa; ETS-GM2: enfermedad de Tay Sachs-gangliosidosis M2; EW: enfermedad de Wilson; ER: enfermedad de Refsum; LCR: líquido cefalorraquídeo; MIRAS: ataxia mitocondrial escandinava de herencia recesiva; NPC: enfermedad de Niemann-Pick tipo C; SANDO: síndrome de ataxia, neuropatía, disartria y oftalmoparesia; SMS: síndrome de Marinesco-Sjögren; XC: xantomatosis cerebrotendinosa.
El examen oftalmológico puede resultar de gran ayuda para sospechar un determinado tipo de heredoataxia recesiva y estos son algunos hallazgos característicos: telangiectasias esclerales en la AT; cataratas en la XC y en el síndrome de Marinesco-Sjögren; anillo de Kayser-Fleischer en la enfermedad de Wilson; fibras mielinizadas en examen de fondo de ojo y aumento de espesor de la capa de fibras en el estudio con tomografía de coherencia óptica en la ARSACS; retinitis pigmentosa en la ABLP y ADVE. La presencia de ictiosis es indicativa de enfermedad de Refsum. En más de la mitad de los pacientes con enfermedad de Niemann-Pick tipo C se encuentra esplenomegalia y muchos de ellos tienen antecedentes de ictericia neonatal prolongada. Los xantomas tendinosos dan nombre a la XC pero pueden estar ausentes. Escoliosis-cardiopatía-diabetes es una tríada que forma parte del espectro clínico de la AF.
Descripción breve de las heredoataxias recesivas más relevantesAtaxia de FriedreichLa AF está causada por mutaciones en el gen FXN (9q21.11), que codifica la síntesis de la frataxina, proteína que interviene en la función mitocondrial activando la fosforilización oxidativa (su falta produce acumulación de hierro mitocondrial y causa estrés oxidativo y muerte celular). En la mayoría de los casos la mutación es una expansión intrónica del triplete GAA en homocigosis18, alrededor del 2% de los casos se producen por una mutación missense en uno de los alelos y una expansión en el otro. La AF es la primera causa de ataxia recesiva en la población caucásica. Su edad de inicio tiene un amplio rango, que puede ir de los 5 a los 25 años, aunque se han descrito casos de inicio tardío (25-39 años) y muy tardío (40 años en adelante)19–21. El fenotipo habitual es el de una ataxia cordonal y cerebelosa progresiva (pérdida de células ganglios sensitivos y degeneración de cordones posteriores y tractos espinocerebelosos), con arreflexia, pie cavo, signo de Babinski y escoliosis, que en 10-15 años confina al paciente en silla de ruedas22. Además de la polineuropatía sensitiva axonal, también es frecuente la neuropatía auditiva y óptica. Miocardiopatía hipertrófica y diabetes son manifestaciones sistémicas relevantes. En los casos de inicio tardío y muy tardío el fenotipo puede ser el de una ataxia con cierto grado de paraparesia, con retención de reflejos tendinosos o un cuadro con similitudes a atrofia multisistémica21 con atrofia cerebelosa y de tronco en el estudio de RM. El tratamiento con idebenona (derivado de la Co-Q10) puede aminorar el desarrollo de miocardiopatía y la progresión del deterioro neurológico17.
Ataxia telangiectasiaEstá causada por mutaciones en el gen ATM (11q22.3)23, que codifica la síntesis de una proteína cinasa implicada en la reparación del ADN: la cantidad/funcionalidad de dicha proteína condiciona la amplia variabilidad fenotípica de la AT24,25. El comienzo de los síntomas suele ser en la primera infancia, aunque hay casos de inicio mucho más tardío. Suele presentarse con ataxia de línea media que progresa a ataxia pancerebelosa, con notable atrofia del cerebelo, hecho que la diferencia de la AF, al igual que el tipo de neuropatía, que es sensitivomotora axonal. Las telangiectasias esclerales y cutáneas son características de la AT, aunque pueden estar ausentes inicialmente. Es frecuente la apraxia óptica y también la coreoatetosis. Los pacientes presentan grados variables de inmunodeficiencia con riesgo de padecer cáncer (leucemias, linfomas). La elevación sérica del antígeno carcinoembrionario y de la alfa-fetoproteína son marcadores constantes, aunque esta última también está elevada en la AOA2, AOA3, AOA4 y la ARCAIII, entidades relacionadas también con alteraciones en la reparación del ADN26. El hallazgo en el cariotipo de traslocaciones 7:14 y la detección de radiosensibilidad aumentada in vitro apoyan el diagnóstico de la AT, que actualmente se basa en la presencia de mutaciones en el gen ATM. La AT carece de tratamiento específico.
Existen otros cuadros denominados AT-like-disorders, uno de ellos el AT-LD1 está causado por mutaciones en gen MRE11A (11q21) que cursa con un cuadro parecido y también radiosensibilidad aumentada pero sin telangiectasias ni inmunodeficiencia27.
Abetalipoproteinemia y ataxia por déficit de vitamina ELa ABLP se produce por mutaciones en el gen MTP (4q23), que codifica la síntesis de la subunidad mayor de la microsomal triglyceride transfer protein, encargada del ensamblaje de la apolipoproteína-B (quilomicrones y proteínas de muy baja densidad28. Suele comenzar en el primer año de vida; su cuadro clínico es parecido al de la AF, pero cursa con un síndrome de malabsorción con hipocolesterolemia y déficits de vitaminas liposolubles (vitamina E incluida), además de acantocitosis y retinitis pigmentosa. El tratamiento dietético y la administración de suplementos vitamínicos mejoran su cuadro clínico.
La ADVE está causada por mutaciones en el gen TTPA (8q12.3), que codifica la síntesis de la transfer alfa-tocoferol protein, encargada de integrar, en el hígado, la vitamina E en las proteínas de muy baja densidad, que la trasportarán al SN. Su fenotipo es similar al de la AF, aunque la disminución de la agudeza visual por retinitis pigmentosa es una característica de la ADVE, en la que suelen faltar la miocardiopatía y la diabetes. También puede cursar con distonía, titubeo (temblor) de cabeza, manifestaciones psiquiátricas y deterioro cognitivo29,30. La determinación de los niveles de vitamina E en cuadros de ataxia no filiados es importante, ya que el tratamiento con dicha vitamina en la ADVE detiene su progresión31.
En la isla Gran Caimán hay un tipo particular de heredoataxia recesiva producida por mutaciones en el gen ATCAY (19p13.3), que también interviene en el metabolismo de la vitamina E.
Ataxias con apraxia oculomotora tipos 1 a 4La AOA1 está causada por mutaciones en el gen APTX (9p21.1), que codifica la síntesis de la aprataxina32, proteína involucrada en la reparación del ADN. La ataxia suele comenzar antes de los 10 años y, a menudo, se acompaña de neuropatía, coreoatetosis, nistagmo y apraxia óptica. Su patrón clínico tiene ciertas similitudes con el de la AT, pero en la AOA1 es frecuente el hallazgo de hipoalbuminemia e hipercolesterolemia, mientras que están ausentes en la AT33. En algunos casos se ha constatado respuesta a la Co-Q1034.
La AOA2 está causada por mutaciones en el gen SETX (9q34.13), que codifica la síntesis de senataxina, helicasa implicada en la transcripción y reparación del ADN, así como el procesado de diversos tipos de ARN35. Su fenotipo es similar al de la AOA1, pero su comienzo es más tardío (segunda década) y no presenta hipoalbuminemia ni hipercolesterolemia, aunque sí elevación de la alfa-fetoproteína36,37, lo que hace necesario un diagnóstico diferencial con la AT. Mutaciones missense del gen SETX en heterocigosis han sido descritas como causa de de esclerosis lateral amiotrófica juvenil tipo 438.
La AOA3 está causada por mutaciones en el gen PIK3R5 (17p13.1). Ha sido descrita en una familia de Arabia Saudita39. Comienza en la segunda década, se acompaña de polineuropatía sensitivomotora axonal. Los niveles de alfa-fetoproteína están elevados.
La AOA4 está causada por mutaciones en el gen PNKP (19q13.33). Ha sido descrita recientemente en Portugal40, donde también la AOA1 es relativamente frecuente. Comienza en la primera década y su evolución es rápida. Cursa con polineuropatía sensitivomotora y algunos casos presentan demencia. También se constató elevación de alfa-fetoproteína en algunos casos.
Ataxia espástica de Charlevoix-SaguenayEstá causada por mutaciones en el gen SACS (13q12.12), que codifica la sacsina, una proteína que tiene actividad de chaperona e interacciona con el proteosoma, pero también interviene en la función mitocondrial y afecta al transporte axonal y dendrítico41. Se cree que la ARSACS es el resultado de un trastorno del desarrollo y de un proceso degenerativo42. Descrita inicialmente en Quebec, posteriormente se han notificado casos en todo el mundo. Su fenotipo es bastante homogéneo y se caracteriza por una ataxia de comienzo temprano, acompañada de piramidalismo, neuropatía sensitivomotora mixta y pie cavo. En algunos casos de inicio tardío, el piramidalismo es más atenuado y hay hipoacusia. En el fondo de ojo pueden observarse persistencia de fibras mielinizadas y aumento del grosor de la capa de fibras retinianas en el estudio de tomografía de coherencia óptica. En el estudio de RM se observa atrofia de la parte posterior del cuerpo calloso, atrofia del vermis cerebeloso, hipointensidades lineales en la protuberancia (fig. 1C y D), y alteración de los tractos pontocerebelosos en la RM-tensor de difusión42,43.
 RM-sagital T1: atrofia intensa del vermis cerebeloso en paciente con ARCA1; B) RM-axial-T2: atrofia pancerebelosa en el mismo paciente con ARCA1. C) RM-axial T1: atrofia de cuerpo calloso posterior en paciente con ARSACS; D) RM-axial T2: hiposeñales lineales protuberanciales en paciente con ARSACS.")
A) RM-sagital T1: atrofia intensa del vermis cerebeloso en paciente con ARCA1; B) RM-axial-T2: atrofia pancerebelosa en el mismo paciente con ARCA1. C) RM-axial T1: atrofia de cuerpo calloso posterior en paciente con ARSACS; D) RM-axial T2: hiposeñales lineales protuberanciales en paciente con ARSACS.
La ARCA1 está causada por mutaciones en el gen SYNE1 (6q25.2) que codifica la síntesis de nesprina-1, una proteína de la membrana nuclear que participa en la ligazón del nucleoesqueleto al citoesquesleto. Suele cursar con un síndrome cerebeloso puro de lenta progresión y determinante de patente atrofia pancerebelosa en el estudio de RM (fig. 1A y B). Su edad media de comienzo se sitúa en torno al final de la tercera década44,45.
La ARCA2 está causada por mutaciones en el gen ADCK3 (1q42.13), que determinan una deficiencia primaria de Co-Q10 y cursa con ataxia cerebelosa de inicio en la primera década; su progresión es lenta y puede asociarse a moderado retraso mental, mioclono, intolerancia al ejercicio, episodios ictus-like y elevación del lactato en sangre46. En el estudio de RM, además de la atrofia cerebelosa, pueden observarse hiperintensidades corticales.
La ARCA3 está causada por mutaciones en el gen ANO10 (3p22.1-p21.3), que da lugar a cuadro de ataxia con piramidalismo y amiotrofia, de comienzo variable (6-45 años) y lenta progresión47. En algunos pacientes se detectó elevación sérica de alfa-fetoproteína.
Enfermedad de RefsumEstá causada por mutaciones en el gen PHYH o PAHX (10p13)48, que codifica una enzima peroxisomal, la fitanol-CoA hidroxilasa, cuyo déficit produce acumulación de ácido fitánico, que daña la mielina. Suele comenzar antes de los 20 años y cursa con ataxia, polineuropatía con hiperproteinorraquia, sordera, retinitis pigmentosa, anosmia, epilepsia y deterioro cognitivo en algunos casos; además suele producir ictiosis, afectación renal y cardiaca, y deformidades esqueléticas49. La RM muestra áreas de hiperseñal no simétricas en cuerpo calloso, haces piramidales y núcleos dentados del cerebelo; las lesiones pueden captar contraste lo que habla a favor de desmielinización activa. La restricción dietética de ácido fitánico y la plasmaféresis, en casos de arritmias cardiacas, mejoran o atenúan su curso progresivo.
Mutaciones en el gen PEX7 (6q23.3), que codifica el receptor peroxina-7, pueden producir un fenotipo similar al del Refsum o cuadros clínicos más graves y complejos como el síndrome de condrodisplasia rizomiélica punctata tipo 1 y otros trastornos como adrenoleucodistrofia neonatal y síndrome de Zellweger50.
Xantomatosis cerebrotendinosaEstá causada por mutaciones en el gen CYP27A1 (2q35), que codifica la enzima 27-esterol-hidroxilasa, que interviene en la síntesis de los ácidos biliares51. Su déficit da lugar a acumulación de diversos esteroles, entre ellos el colestanol, que se encuentra elevado en el suero. El cuadro neurológico suele ser notorio al final de la segunda década y combina epilepsia, ataxia, piramidalismo y polineuropatía. También son frecuentes los trastornos psiquiátricos. Antes del inicio del cuadro neurológico son frecuentes las diarreas, las cataratas y la presencia de xantomas tendinosos, aunque hemos estudiado a 3 pacientes de 2 familias distintas, en los que este signo que da nombre a la enfermedad estaba ausente; este hecho provocó retrasos en el diagnóstico52,53. En la RM pueden observarse, en algunos casos, hiperintensidades en núcleos dentados; también senos craneales grandes y atrofia de médula cervical (fig. 2B). El tratamiento con ácido quenodesoxicólico enlentece la progresión de la XC.
 RM-axial T2: hiperintensidad periacueductal y de calota mesencefálica contrastando con la hipointensidad de cuerpos mamilares, núcleos rojos y sustancia negra perfilando «cara de oso panda» en paciente con EW. B) RM-sagital T1: senos craneales prominentes y atrofia medular en paciente con XC. C) RM-T2: atrofia cortical, de cuerpo calloso, tronco y vermis cerebeloso en paciente joven con enfermedad de NPC.")
Heredoataxias metabólicas tratables: A) RM-axial T2: hiperintensidad periacueductal y de calota mesencefálica contrastando con la hipointensidad de cuerpos mamilares, núcleos rojos y sustancia negra perfilando «cara de oso panda» en paciente con EW. B) RM-sagital T1: senos craneales prominentes y atrofia medular en paciente con XC. C) RM-T2: atrofia cortical, de cuerpo calloso, tronco y vermis cerebeloso en paciente joven con enfermedad de NPC.
Se produce por mutaciones en los genes NPC1 (18q11.2) en el 90% de los casos y NPC2 (14q24.3) en el 10% restante; el primero codifica una proteína transmembrana y el segundo una intralisosomal, cuyos déficits distorsionan el tráfico de colesterol, esfingosina y glucolípidos, que se acumulan en los lisosomas y determinan muerte celular54. La enfermedad puede tener distintos fenotipos clínicos, que dependen de la edad de comienzo: forma neonatal fatal, forma infantil precoz con hipotonía y retraso psicomotor, forma infantil tardía (torpeza, trastornos de la marcha y de adquisición del lenguaje, cataplejía gelástica), forma juvenil (ataxia, epilepsia, cataplejía, mal rendimiento escolar) y forma adulta (trastorno psiquiátrico, ataxia, distonía, demencia)55. Ahora con las técnicas de secuenciación masiva se descubren más casos tardíos56. La parálisis de la mirada vertical suele estar presente en las 3 últimas formas. Es frecuente el antecedente de ictericia neonatal prolongada y la presencia de esplenomegalia. En la RM se observa atrofia cerebral, de tronco y cerebelo (fig. 2C). La actividad de la quitiotriosidasa macrofágica y la CCL18 plasmática no siempre están elevadas. La tinción con filipina de cultivo de fibroblastos demuestra la acumulación de colesterol. El tratamiento con miglustat puede mejorar la evolución.
Enfermedad de WilsonEstá causada por mutaciones en el gen ATP7B (13q14.3), que codifica la síntesis de una ATPasa que participa en el transporte del cobre a través del aparato de Golgi del hepatocito57. Puede tener diversas formas de presentación, entre ellas hepatitis fulminante, psicosis o trastorno neurológico (temblor, distonía, síndrome rígido-acinético y ocasionalmente ataxia). El anillo corneal de Kayser-Fleischer se encuentra en la mayoría de los pacientes con clínica neurológica58. Los hallazgos en el estudio de RM (fig. 2A), tal como se describió anteriormente, son muy variables59. En sangre se detectan niveles bajos de ceruloplasmina y de cobre, cuya excreción está aumentada en orina. La D-penicilamina y trientine como quelantes, y el tetratiomolibdato y sulfato de cinc constituyen el tratamiento habitual59.
Síndrome de Marinesco-SjögrenEstá causado por mutaciones en el gen SIL1 (5q31.2), que codifica la chaperona HSPA5, un factor de recambio para la heat-shock-protein-70 (HSP-70)60. Da lugar a talla corta, hipogonadismo hipergonadotrófico, deformidades esqueléticas, miopatía, cataratas congénitas y ataxia cerebelosa, con gran atrofia en el estudio de RM (fig. 3).
Enfermedad de Tay-Sachs (gangliosidosis GM2) RM-sagital T1: atrofia de tronco y extensa atrofia vermiana. B) RM-axial T2: atrofia protuberancial, vermiana y de hemisferios cerebelosos, sin alteraciones de señal intraparenquimatosas.")
Está causada por deficiencia de la enzima beta-hexosaminidasa, codificada por el gen HEXA (15q23), que afecta sobre todo a judíos Ashkenazis61. Tiene varias formas de inicio: la infantil cursa con hipotonía, ceguera (manchas rojo-cereza en fondo de ojo), retraso mental grave y muerte alrededor de los 3 años; en formas de inicio tardío (juvenil, adulto) puede dar lugar a ataxia, enfermedad de motoneurona inferior con atrofia y fasciculaciones, espasticidad, demencia y epilepsia62.
Heredoataxias mitocondriales recesivasMutaciones en el gen POLG(1) (15q26.1), que codifica una subunidad de una polimerasa reparadora del ADN mitocondrial, pueden dar lugar a múltiples fenotipos, con distintos patrones de herencia: entre ellos, un síndrome de ataxia de inicio infantil o en adultos jóvenes, asociada a neuropatía sensitiva, disartria y oftalmoplejía, y en algunos casos acidosis láctica, epilepsia, miopatía con fibras rojas hendidas y hepatopatía. En el estudio de RM pueden observarse hiperintensidades talámicas y atrofia cerebelosa63. Estos trastornos pueden ser de herencia recesiva o dominante y son causa de deleciones secundarias del ADN mitocondrial. El denominado síndrome de ataxia mitocondrial recesiva escandinávica está causado por las mutaciones A467T y W748S en POLG y constituye una causa frecuente de ataxia en Finlandia y Noruega, y también otros países de Europa Central64. Hay mutaciones en otros genes, relacionados con la función mitocondrial, como el RRM2B (8q22.3) que pueden dar lugar a cuadros de ataxia65.
La ataxia espinocerebelosa de inicio infantil está causada por mutaciones en homo- y heterocigosis de C10orf2 (10q23.3-24.1), que codifica las proteínas twinkle y twinky, y da lugar al síndrome de depleción mitocondrial tipo 7, que puede cursar con oftalmoplejía, sordera, ataxia espinocerebelosa temprana y epilepsia refractaria66. Es la segunda causa de ataxia en Finlandia.
ConclusionesLos fenotipos clínicos de las heredoataxias recesivas son muy variables y a menudo se solapan. El clínico puede tener la sensación de verse perdido en un laberinto14 y la salida del mismo puede parecerle un reto imposible. El conocimiento de la clínica y los marcadores (laboratorio, electromiografía y neuroimagen) de las entidades principales (resumidos en la figura 4) facilita la labor y, de este modo, en bastantes casos no habrá que recurrir a las técnicas de secuenciación masiva (exomas) ni a los extensos y todavía costosos paneles de genes. Sin el conocimiento de la mutación genética no se puede establecer un pronóstico ni un adecuado consejo genético, ni mucho menos atisbar un tratamiento satisfactorio. Aunque ya se conoce que defectos en la reparación del ADN, disfunción mitocondrial, trastornos de la transmisión sináptica, pérdida de la actividad de la función de chaperonas y alteraciones de rutas metabólicas son mecanismos fisiopatológicos implicados en la génesis de las heredoataxias recesivas67, pocas tienen un tratamiento efectivo y lo más prioritario es diagnosticarlas cuanto antes.

Cuatro pilares para afrontar el diagnóstico de un síndrome cerebeloso de probable naturaleza degenerativa y con patrón de herencia autosómica recesiva. ABLP: abetalipoproteinemia; ADVE: ataxia por déficit de vitamina E; AF: ataxia de Friedreich; AT: ataxia telangiectasia; AOAI: ataxia con apraxia oculomotora; ARCA: ataxia recesiva cerebelosa; ARSACS: ataxia recesiva de Charlevoix-Saguenay; ER: enfermedad de Refsum; ETS-GM2: enfermedad de Tay Sachs-gangliosidosis M2; EW: enfermedad de Wilson; MIRAS: ataxia mitocondrial escandinava de herencia recesiva; NPC: enfermedad de Niemann-Pick tipo C; SANDO: síndrome de ataxia, neuropatía, disartria y oftalmoparesia; SMS: síndrome de Marinesco-Sjögren; XC: xantomatosis cerebrotendinosa.
No.
A los Dres. Ángel Ortega (Granada) y Juan Manuel Pías-Peleteiro (Santiago de Compostela) por cederme algunas de las imágenes.

