




Global developmental delay (GDD) and intellectual disability (ID) are frequent reasons for consultation in paediatric neurology departments. Nowadays, array comparative genomic hybridisation (array-CGH) is one of the most widely used techniques for diagnosing these disorders. Our purpose was to determine the phenotypic features associated with pathological results in this genetic test.
MethodsWe conducted a blind study of the epidemiological, clinical, anthropometric, and morphological features of 80 patients with unexplained ID to determine which features were associated with pathological results in array-CGH.
ResultsPathological results were found in 27.5% of the patients. Factors associated with pathological results in array-CGH were a family history of GDD/ID (OR=12.1), congenital malformations (OR=5.33), having more than 3 facial dysmorphic features (OR=20.9), and hypotonia (OR=3.25).
ConclusionsOur findings are consistent with those reported by other published series. We therefore conclude that the probability of having pathological results in array-CGH increases with the presence of any of the features mentioned above in patients with ID/GDD.
El retraso global del desarrollo (RGD) y la discapacidad intelectual (DI) son un motivo de consulta frecuente en la consulta de Neuropediatría. Actualmente, la hibridación genómica comparada constituye una de las principales técnicas aplicadas al diagnóstico de esta patología. Resulta útil determinar qué características fenotípicas se asocian a obtener un resultado etiológico en el test genético.
MétodosSe llevó a cabo un estudio ciego pormenorizado de las características clínicas, antropométricas y morfológicas de 80 individuos afectos de DI no explicada y se analizó cuales estaban asociadas a obtener un resultado etiológico en el array-CGH.
ResultadosEl resultado del array fue patológico en un 27,5% de los casos. Los factores que se asociaron estadísticamente a tener una prueba de array-CGH patológica fueron los antecedentes familiares de DI/RGD (OR: 12,1), la presencia de malformaciones congénitas (OR: 5,33), más de 3 rasgos dismórficos faciales (OR: 20,9) y la hipotonía periférica (OR: 3,25).
ConclusionesNuestros hallazgos coinciden con otras series publicadas. Por lo tanto, asumimos que la probabilidad de encontrar variación en el número de copias de significado patológico mediante array-CGH aumenta si alguna de las características anteriores está presente en individuos afectos de DI/RGD.
Intellectual disability (ID) is characterised by significant limitations in intellectual and adaptive functioning and originates before the age of 18 years. Children younger than 5 who have not achieved the social, motor, or intellectual developmental milestones expected for their age (especially language) are described as having global developmental delay (GDD).1–3 The aetiological diagnosis of ID/GDD is unknown in approximately 50% of cases.4,5 The most frequently identified genetic causes are point mutations in known genes and chromosomal alterations.6,7 Array comparative genomic hybridisation (CGH) identifies duplications and deletions of genetic material and constitutes one of the main diagnostic tests for children with neurodevelopmental disorders. For ID, the diagnostic yield of this technique ranges from 10% to 30%, depending on the selection criteria of the sample.8,9 Our objective was to determine the most frequent clinical, anthropometric, and morphological characteristics in patients with ID/GDD and pathological array CGH results.
Patients and methodsParticipants had to meet the following inclusion criteria: (1) being followed up by the paediatric neurology department at Hospital Infantil Miguel Servet in Zaragoza, Spain; (2) being aged between 12 months and 18 years; (3) having ID or GDD with no established aetiological diagnosis; and (4) having undergone array CGH and received the results before 1 December 2011. A total of 140 patients met our inclusion criteria. The study spanned a period of 2 years and was developed by a single researcher.
We established a diagnosis of GDD for those patients younger than 5 years with neurodevelopmental alterations in 2 or more categories of the Denver Developmental Screening Test or Haizea-Llevant scale. Likewise, those children aged≥5 years with an IQ<70 were diagnosed with ID. When no neuropsychological study was available, presence of ID was established based on type of schooling (special education or significant curriculum adaptation).
The families of the patients were informed in writing about the characteristics of the study; in-person appointments were scheduled by phone. Informed consent forms were signed on the day of the appointment. The main demographic, perinatal, clinical, morphological, and anthropometric data for each participant were gathered through clinical interviews and thorough physical examinations. Clinical histories and array CGH results were reviewed subsequently; results were classified as normal, pathological, or uncertain. This classification was based on data from the following databases: the Database of Genomic Variants (DGV), the International Standards for Cytogenomic Arrays (ISCA) Consortium databases, the Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources (DECIPHER), and the Online Mendelian Inheritance in Man (OMIM) database.
This study complies with the ethical principles of the Declaration of Helsinki (59th WMA General Assembly, Seoul, Republic of Korea, October 2008).10 The project was reviewed and approved by the clinical research ethics committee of the region of Aragón. All patients signed informed consent forms.
Genetic testing was conducted on DNA obtained from peripheral blood lymphocytes using the EZ1 automated genomic DNA purification system (QUIAGEN GmbH, Hilden, Germany). Samples were sent to 2 different laboratories for array CGH: qGenomics and Genycell Biotech. Hybridisation was conducted using an array comprising 60000 oligonucleotide probes from loci across the entire genome, with greater coverage at pericentromeric and subtelomeric regions and those involved in recurrent genetic disorders.
After the variables had been collected, we checked for normal distribution and homogeneity of variance using the Shapiro–Wilk test and the Levene test, respectively. The association between the factors studied and the array CGH results was analysed using hypothesis contrast testing, comparing proportions when both variables were qualitative (chi-square test for normally distributed data and Fisher exact test for non-normally distributed data) and means when one of them was quantitative (t test and ANOVA for normal distributions and Mann–Whitney U test for non-normal distributions). We compared the distribution of characteristics between patients with normal and pathological array CGH results. We excluded participants with uncertain array CGH results and patient no. 15, who also had fragile X syndrome and whose characteristics may have acted as confounding factors.
In the logistic regression model, univariate regression analysis was used to screen for explanatory variables, selecting the potential prognostic factors that may be included in the multivariate analysis (P<.25).11,12 The resulting variables and those that had to be included in the final model for clinical reasons were included in different multivariate models using forward selection and backward elimination; P values of .1 and .2 were used for inclusion or exclusion of variables, respectively. In all cases, the statistical significance level was set at P<.05. Statistical analysis was performed with IBM SPSS Statistics version 21 (SPSS Inc., Chicago, USA).
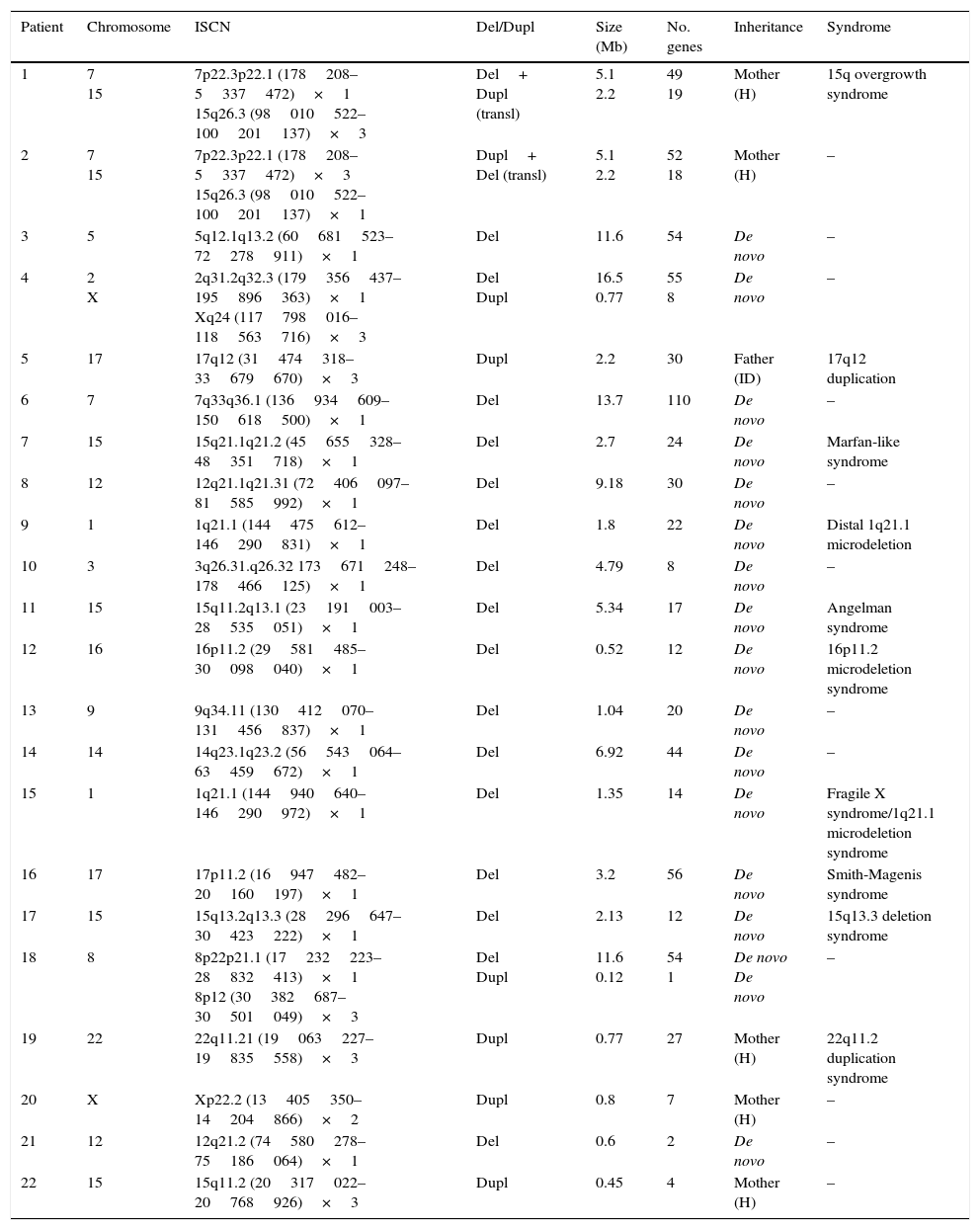
ResultsEighty patients completed all the phases of the study (Fig. 1). We found copy number variants (CNV) in 40% of cases; 27.5% were pathological (Table 1). Regarding the type of chromosome imbalance, 62.5% of the cases were deletions, 6.25% were unbalanced translocations, and 31.25% were duplications. Deletions ranged between 0.06 and 16.5Mb in size and duplications between 0.08 and 2.2Mb. De novo chromosomal alterations were found in 51.6% of analyses. Chromosomal alterations inherited from a single parent were from the father in 7 cases (one affected father, the remaining 6 were healthy) and the mother in 8 (all mothers were healthy, although 2 were balanced translocation carriers).

Genetic characteristics of patients with pathological array CGH results.
| Patient | Chromosome | ISCN | Del/Dupl | Size (Mb) | No. genes | Inheritance | Syndrome |
|---|---|---|---|---|---|---|---|
| 1 | 7 15 | 7p22.3p22.1 (178208–5337472)×1 15q26.3 (98010522–100201137)×3 | Del+ Dupl (transl) | 5.1 2.2 | 49 19 | Mother (H) | 15q overgrowth syndrome |
| 2 | 7 15 | 7p22.3p22.1 (178208–5337472)×3 15q26.3 (98010522–100201137)×1 | Dupl+ Del (transl) | 5.1 2.2 | 52 18 | Mother (H) | – |
| 3 | 5 | 5q12.1q13.2 (60681523–72278911)×1 | Del | 11.6 | 54 | De novo | – |
| 4 | 2 X | 2q31.2q32.3 (179356437–195896363)×1 Xq24 (117798016–118563716)×3 | Del Dupl | 16.5 0.77 | 55 8 | De novo | – |
| 5 | 17 | 17q12 (31474318–33679670)×3 | Dupl | 2.2 | 30 | Father (ID) | 17q12 duplication |
| 6 | 7 | 7q33q36.1 (136934609–150618500)×1 | Del | 13.7 | 110 | De novo | – |
| 7 | 15 | 15q21.1q21.2 (45655328–48351718)×1 | Del | 2.7 | 24 | De novo | Marfan-like syndrome |
| 8 | 12 | 12q21.1q21.31 (72406097–81585992)×1 | Del | 9.18 | 30 | De novo | – |
| 9 | 1 | 1q21.1 (144475612–146290831)×1 | Del | 1.8 | 22 | De novo | Distal 1q21.1 microdeletion |
| 10 | 3 | 3q26.31.q26.32 173671248–178466125)×1 | Del | 4.79 | 8 | De novo | – |
| 11 | 15 | 15q11.2q13.1 (23191003–28535051)×1 | Del | 5.34 | 17 | De novo | Angelman syndrome |
| 12 | 16 | 16p11.2 (29581485–30098040)×1 | Del | 0.52 | 12 | De novo | 16p11.2 microdeletion syndrome |
| 13 | 9 | 9q34.11 (130412070–131456837)×1 | Del | 1.04 | 20 | De novo | – |
| 14 | 14 | 14q23.1q23.2 (56543064–63459672)×1 | Del | 6.92 | 44 | De novo | – |
| 15 | 1 | 1q21.1 (144940640–146290972)×1 | Del | 1.35 | 14 | De novo | Fragile X syndrome/1q21.1 microdeletion syndrome |
| 16 | 17 | 17p11.2 (16947482–20160197)×1 | Del | 3.2 | 56 | De novo | Smith-Magenis syndrome |
| 17 | 15 | 15q13.2q13.3 (28296647–30423222)×1 | Del | 2.13 | 12 | De novo | 15q13.3 deletion syndrome |
| 18 | 8 | 8p22p21.1 (17232223–28832413)×1 8p12 (30382687–30501049)×3 | Del Dupl | 11.6 0.12 | 54 1 | De novo De novo | – |
| 19 | 22 | 22q11.21 (19063227–19835558)×3 | Dupl | 0.77 | 27 | Mother (H) | 22q11.2 duplication syndrome |
| 20 | X | Xp22.2 (13405350–14204866)×2 | Dupl | 0.8 | 7 | Mother (H) | – |
| 21 | 12 | 12q21.2 (74580278–75186064)×1 | Del | 0.6 | 2 | De novo | – |
| 22 | 15 | 15q11.2 (20317022–20768926)×3 | Dupl | 0.45 | 4 | Mother (H) | – |
Del, deletion; ID, intellectual disability; Dupl, duplication; ISCN, International System for Human Cytogenomic Nomenclature; H, healthy; transl, translocation.
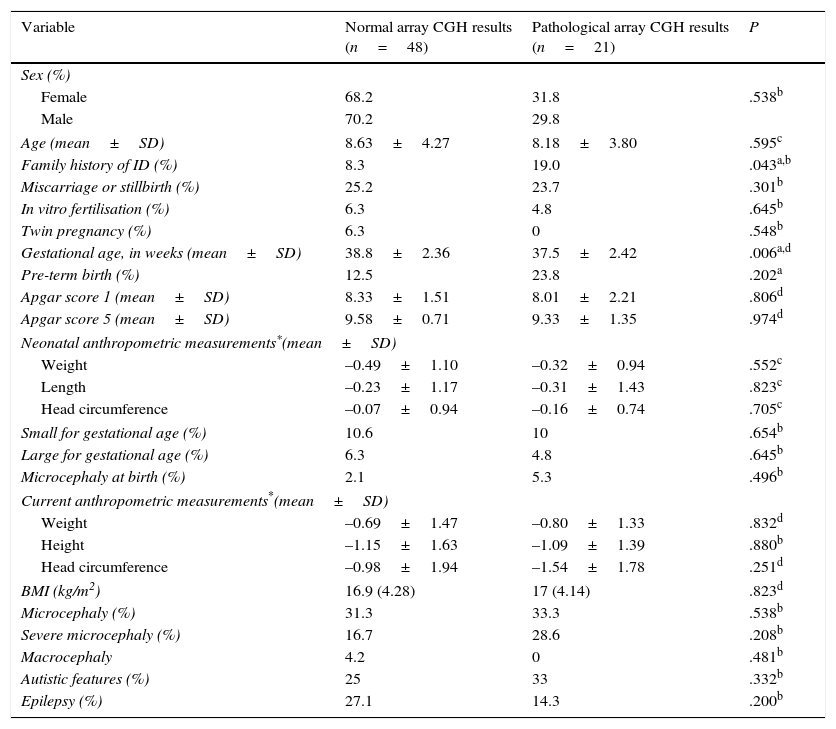
Regarding the main demographic, perinatal, anthropometric, and clinical data, we found an association between family history of ID and pathological array CGH results (P=.043; OR 12.1; 95% CI, 3.18-45.9). Mean gestational age was lower among patients with pathological array CGH results (P=.006), although no significant differences were found in the rate of premature birth between groups (Table 2).
Comparison of demographic, perinatal, anthropometric, and clinical characteristics between patients with and without pathological array CGH results.
| Variable | Normal array CGH results (n=48) | Pathological array CGH results (n=21) | P |
|---|---|---|---|
| Sex (%) | |||
| Female | 68.2 | 31.8 | .538b |
| Male | 70.2 | 29.8 | |
| Age (mean±SD) | 8.63±4.27 | 8.18±3.80 | .595c |
| Family history of ID (%) | 8.3 | 19.0 | .043a,b |
| Miscarriage or stillbirth (%) | 25.2 | 23.7 | .301b |
| In vitro fertilisation (%) | 6.3 | 4.8 | .645b |
| Twin pregnancy (%) | 6.3 | 0 | .548b |
| Gestational age, in weeks (mean±SD) | 38.8±2.36 | 37.5±2.42 | .006a,d |
| Pre-term birth (%) | 12.5 | 23.8 | .202a |
| Apgar score 1 (mean±SD) | 8.33±1.51 | 8.01±2.21 | .806d |
| Apgar score 5 (mean±SD) | 9.58±0.71 | 9.33±1.35 | .974d |
| Neonatal anthropometric measurements*(mean±SD) | |||
| Weight | –0.49±1.10 | –0.32±0.94 | .552c |
| Length | –0.23±1.17 | –0.31±1.43 | .823c |
| Head circumference | –0.07±0.94 | –0.16±0.74 | .705c |
| Small for gestational age (%) | 10.6 | 10 | .654b |
| Large for gestational age (%) | 6.3 | 4.8 | .645b |
| Microcephaly at birth (%) | 2.1 | 5.3 | .496b |
| Current anthropometric measurements*(mean±SD) | |||
| Weight | –0.69±1.47 | –0.80±1.33 | .832d |
| Height | –1.15±1.63 | –1.09±1.39 | .880b |
| Head circumference | –0.98±1.94 | –1.54±1.78 | .251d |
| BMI (kg/m2) | 16.9 (4.28) | 17 (4.14) | .823d |
| Microcephaly (%) | 31.3 | 33.3 | .538b |
| Severe microcephaly (%) | 16.7 | 28.6 | .208b |
| Macrocephaly | 4.2 | 0 | .481b |
| Autistic features (%) | 25 | 33 | .332b |
| Epilepsy (%) | 27.1 | 14.3 | .200b |
BMI, body mass index (kg/m2).
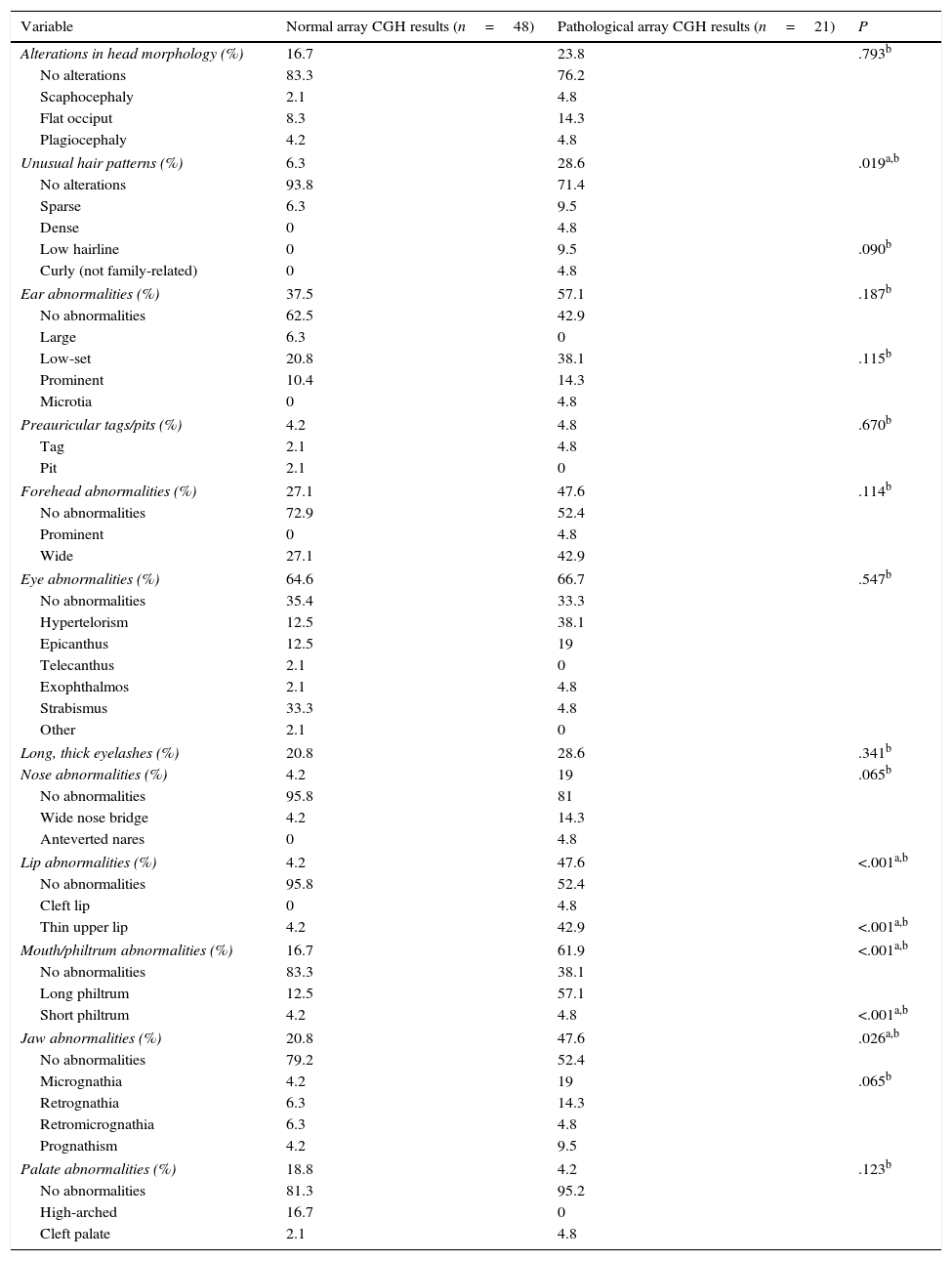
The presence of dysmorphic craniofacial features was analysed in 2 ways: by comparing presence or absence of any type of alteration in each category (eyes, ears, jaw, etc.) using the Fisher exact test, and by comparing the types of alterations detected in each category with the chi-square test (Table 3).
Comparison of dysmorphic craniofacial features between patients with and without pathological array CGH results.
| Variable | Normal array CGH results (n=48) | Pathological array CGH results (n=21) | P |
|---|---|---|---|
| Alterations in head morphology (%) | 16.7 | 23.8 | .793b |
| No alterations | 83.3 | 76.2 | |
| Scaphocephaly | 2.1 | 4.8 | |
| Flat occiput | 8.3 | 14.3 | |
| Plagiocephaly | 4.2 | 4.8 | |
| Unusual hair patterns (%) | 6.3 | 28.6 | .019a,b |
| No alterations | 93.8 | 71.4 | |
| Sparse | 6.3 | 9.5 | |
| Dense | 0 | 4.8 | |
| Low hairline | 0 | 9.5 | .090b |
| Curly (not family-related) | 0 | 4.8 | |
| Ear abnormalities (%) | 37.5 | 57.1 | .187b |
| No abnormalities | 62.5 | 42.9 | |
| Large | 6.3 | 0 | |
| Low-set | 20.8 | 38.1 | .115b |
| Prominent | 10.4 | 14.3 | |
| Microtia | 0 | 4.8 | |
| Preauricular tags/pits (%) | 4.2 | 4.8 | .670b |
| Tag | 2.1 | 4.8 | |
| Pit | 2.1 | 0 | |
| Forehead abnormalities (%) | 27.1 | 47.6 | .114b |
| No abnormalities | 72.9 | 52.4 | |
| Prominent | 0 | 4.8 | |
| Wide | 27.1 | 42.9 | |
| Eye abnormalities (%) | 64.6 | 66.7 | .547b |
| No abnormalities | 35.4 | 33.3 | |
| Hypertelorism | 12.5 | 38.1 | |
| Epicanthus | 12.5 | 19 | |
| Telecanthus | 2.1 | 0 | |
| Exophthalmos | 2.1 | 4.8 | |
| Strabismus | 33.3 | 4.8 | |
| Other | 2.1 | 0 | |
| Long, thick eyelashes (%) | 20.8 | 28.6 | .341b |
| Nose abnormalities (%) | 4.2 | 19 | .065b |
| No abnormalities | 95.8 | 81 | |
| Wide nose bridge | 4.2 | 14.3 | |
| Anteverted nares | 0 | 4.8 | |
| Lip abnormalities (%) | 4.2 | 47.6 | <.001a,b |
| No abnormalities | 95.8 | 52.4 | |
| Cleft lip | 0 | 4.8 | |
| Thin upper lip | 4.2 | 42.9 | <.001a,b |
| Mouth/philtrum abnormalities (%) | 16.7 | 61.9 | <.001a,b |
| No abnormalities | 83.3 | 38.1 | |
| Long philtrum | 12.5 | 57.1 | |
| Short philtrum | 4.2 | 4.8 | <.001a,b |
| Jaw abnormalities (%) | 20.8 | 47.6 | .026a,b |
| No abnormalities | 79.2 | 52.4 | |
| Micrognathia | 4.2 | 19 | .065b |
| Retrognathia | 6.3 | 14.3 | |
| Retromicrognathia | 6.3 | 4.8 | |
| Prognathism | 4.2 | 9.5 | |
| Palate abnormalities (%) | 18.8 | 4.2 | .123b |
| No abnormalities | 81.3 | 95.2 | |
| High-arched | 16.7 | 0 | |
| Cleft palate | 2.1 | 4.8 | |
Patients with pathological array CGH results displayed more dysmorphic features in all categories except for palate abnormalities. More specifically, this group of patients displayed a significantly higher prevalence of dysmorphic features (P<.05) in the following categories: hair (P=.019), with a higher proportion of patients with unusual hair patterns (OR 6; 95% CI, 1.33-26.9); lips (P<.001), with a high proportion of patients with thin upper lip (P<.001; OR 20.6; 95% CI, 3.99-109.37); mouth and philtrum (P<.001; OR 8.13; 95% CI, 2.54-25.9), with a high proportion of patients with long philtrum (P<.001); and jaw (P=.026; OR 3.45; 95% CI, 1.15-10.42), with micrognathia being the most frequent characteristic (nearly significant, at P=.065) (Table 3). Some 68.4% of patients with pathological array CGH results had more than 3 dysmorphic features, compared to 31.2% of those with normal results (P<.001; OR 20.9; 95% CI, 4.31-101.4).
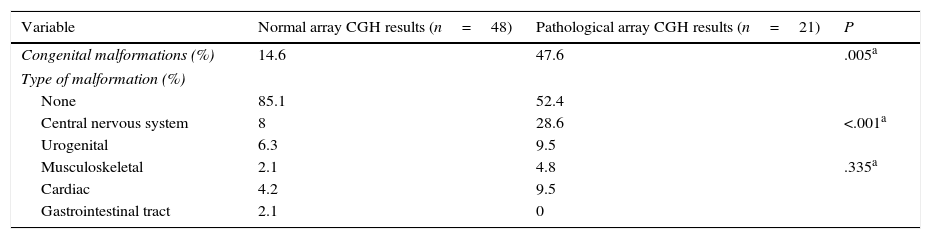
Congenital malformations were more frequent in the group displaying pathological results (P=.005), both in general terms and in each category, with the exception of gastrointestinal tract abnormalities. The most frequent type of congenital alterations in our sample involved the central nervous system (including spinal cord abnormalities); cardiac and urogenital abnormalities were also common, although differences between groups were not significant (Table 4).
Congenital malformations in patients with normal and pathological array CGH results.
| Variable | Normal array CGH results (n=48) | Pathological array CGH results (n=21) | P |
|---|---|---|---|
| Congenital malformations (%) | 14.6 | 47.6 | .005a |
| Type of malformation (%) | |||
| None | 85.1 | 52.4 | |
| Central nervous system | 8 | 28.6 | <.001a |
| Urogenital | 6.3 | 9.5 | |
| Musculoskeletal | 2.1 | 4.8 | .335a |
| Cardiac | 4.2 | 9.5 | |
| Gastrointestinal tract | 2.1 | 0 | |
Statistical analysis.
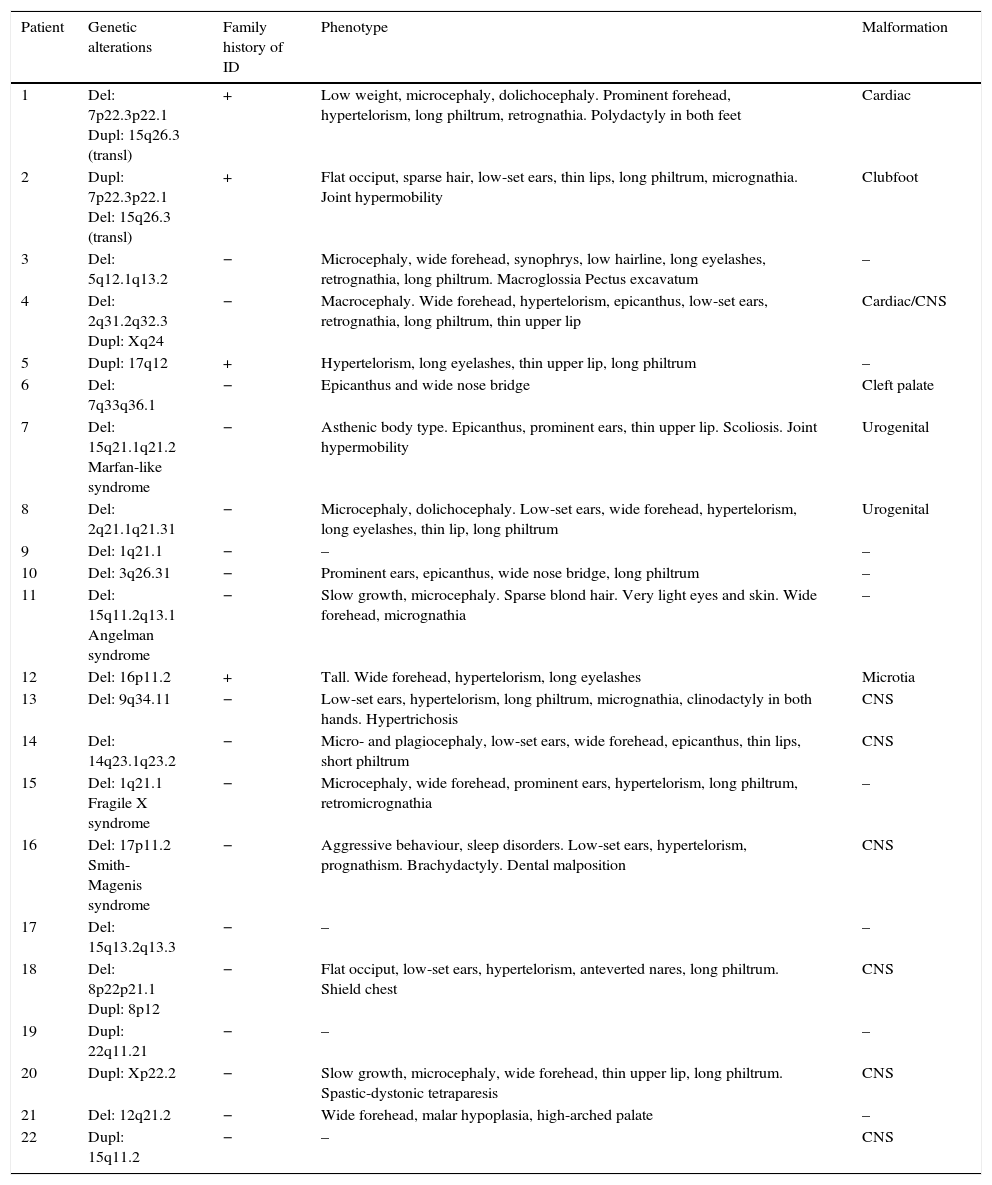
Table 5 shows the main dysmorphic features, presence of congenital malformations, and family history of ID in each of the patients with pathological array CGH results.
Main characteristics of patients with pathological array CGH results.
| Patient | Genetic alterations | Family history of ID | Phenotype | Malformation |
|---|---|---|---|---|
| 1 | Del: 7p22.3p22.1 Dupl: 15q26.3 (transl) | + | Low weight, microcephaly, dolichocephaly. Prominent forehead, hypertelorism, long philtrum, retrognathia. Polydactyly in both feet | Cardiac |
| 2 | Dupl: 7p22.3p22.1 Del: 15q26.3 (transl) | + | Flat occiput, sparse hair, low-set ears, thin lips, long philtrum, micrognathia. Joint hypermobility | Clubfoot |
| 3 | Del: 5q12.1q13.2 | − | Microcephaly, wide forehead, synophrys, low hairline, long eyelashes, retrognathia, long philtrum. Macroglossia Pectus excavatum | – |
| 4 | Del: 2q31.2q32.3 Dupl: Xq24 | − | Macrocephaly. Wide forehead, hypertelorism, epicanthus, low-set ears, retrognathia, long philtrum, thin upper lip | Cardiac/CNS |
| 5 | Dupl: 17q12 | + | Hypertelorism, long eyelashes, thin upper lip, long philtrum | – |
| 6 | Del: 7q33q36.1 | − | Epicanthus and wide nose bridge | Cleft palate |
| 7 | Del: 15q21.1q21.2 Marfan-like syndrome | − | Asthenic body type. Epicanthus, prominent ears, thin upper lip. Scoliosis. Joint hypermobility | Urogenital |
| 8 | Del: 2q21.1q21.31 | − | Microcephaly, dolichocephaly. Low-set ears, wide forehead, hypertelorism, long eyelashes, thin lip, long philtrum | Urogenital |
| 9 | Del: 1q21.1 | − | – | – |
| 10 | Del: 3q26.31 | − | Prominent ears, epicanthus, wide nose bridge, long philtrum | – |
| 11 | Del: 15q11.2q13.1 Angelman syndrome | − | Slow growth, microcephaly. Sparse blond hair. Very light eyes and skin. Wide forehead, micrognathia | – |
| 12 | Del: 16p11.2 | + | Tall. Wide forehead, hypertelorism, long eyelashes | Microtia |
| 13 | Del: 9q34.11 | − | Low-set ears, hypertelorism, long philtrum, micrognathia, clinodactyly in both hands. Hypertrichosis | CNS |
| 14 | Del: 14q23.1q23.2 | − | Micro- and plagiocephaly, low-set ears, wide forehead, epicanthus, thin lips, short philtrum | CNS |
| 15 | Del: 1q21.1 Fragile X syndrome | − | Microcephaly, wide forehead, prominent ears, hypertelorism, long philtrum, retromicrognathia | – |
| 16 | Del: 17p11.2 Smith-Magenis syndrome | − | Aggressive behaviour, sleep disorders. Low-set ears, hypertelorism, prognathism. Brachydactyly. Dental malposition | CNS |
| 17 | Del: 15q13.2q13.3 | − | – | – |
| 18 | Del: 8p22p21.1 Dupl: 8p12 | − | Flat occiput, low-set ears, hypertelorism, anteverted nares, long philtrum. Shield chest | CNS |
| 19 | Dupl: 22q11.21 | − | – | – |
| 20 | Dupl: Xp22.2 | − | Slow growth, microcephaly, wide forehead, thin upper lip, long philtrum. Spastic-dystonic tetraparesis | CNS |
| 21 | Del: 12q21.2 | − | Wide forehead, malar hypoplasia, high-arched palate | – |
| 22 | Dupl: 15q11.2 | − | – | CNS |
Del, deletion; Dupl, duplication; ID, intellectual disability; transl, translocation; CNS, central nervous system.
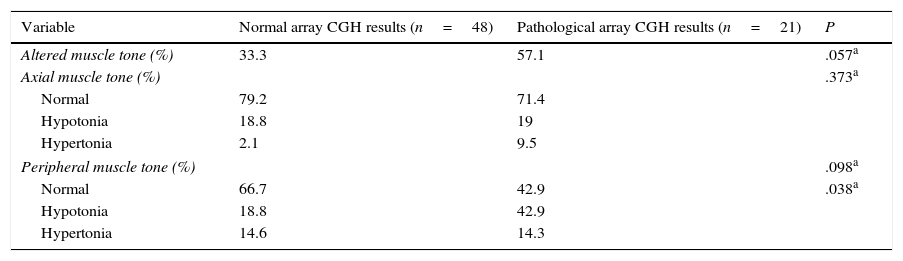
Regarding muscle tone, hypotonia was more frequent than hypertonia at both the axial and peripheral levels. The percentage of patients with axial hypotonia was similar in both groups, whereas peripheral hypotonia was more frequent among patients with pathological array CGH results (Table 6).
Axial and peripheral muscle tone in our sample.
| Variable | Normal array CGH results (n=48) | Pathological array CGH results (n=21) | P |
|---|---|---|---|
| Altered muscle tone (%) | 33.3 | 57.1 | .057a |
| Axial muscle tone (%) | .373a | ||
| Normal | 79.2 | 71.4 | |
| Hypotonia | 18.8 | 19 | |
| Hypertonia | 2.1 | 9.5 | |
| Peripheral muscle tone (%) | .098a | ||
| Normal | 66.7 | 42.9 | .038a |
| Hypotonia | 18.8 | 42.9 | |
| Hypertonia | 14.6 | 14.3 | |
Statistical analysis.
Through multivariate logistic regression analysis, we identified the main factors independently associated with pathological array CGH results: a family history of ID, more than 3 dysmorphic craniofacial features, and lip abnormalities.
The results of genetic tests conducted previously in the group showing pathological array CGH results were not conclusive. Fifteen patients were studied by karyotyping (high-resolution karyotyping in 4) and a study of subtelomeric deletion was performed in 5. Fragile X syndrome was ruled out in 14 boys and 2 girls. The case of patient 15 is of particular interest: fragile X syndrome was ruled out by Southern blot in 2006. However, as the patient's phenotype was compatible with the disease (except for microcephaly), TP-PCR was performed in 2012, revealing over 250 CGG trinucleotide repeats.
DiscussionArray CGH has provided significant advances in the study of ID/GDD and is regarded by many authors as the diagnostic technique of choice for these patients.13,14
One limitation of the study is the small sample size. We should stress, however, that patient characteristics were studied by a single researcher blinded to array CGH results, which minimises variability and increases internal validity.
The number of patients with pathological array CGH results is higher in our sample than in most published series, with rates ranging from 5% to 25%.13,14 This may be due to the inclusion criteria for genetic testing: patients with a family history of ID, dysmorphic features, and/or congenital malformations. Published series using similar inclusion criteria to our own report similar detection rates.8,15
Sixty percent of the alterations detected were deletions. There are 2 possible explanations for this. From a technical viewpoint, duplications are more likely to go undetected. From a biological viewpoint, duplications have been observed to cause less severe phenotypes, which may have led to a loss of cases in the process of sample selection. Furthermore, duplications are less likely to occur at random in the human genome than deletions.14
CNVs do not seem to be distributed at random; rather, these variants tend to accumulate in certain ‘hotspots’. These hotspots usually contain high numbers of genes and regions of segmental duplications.16 In our sample, most CNVs affect genes involved in cell signalling and adhesion or which code for structural proteins of the central nervous system.17
Regarding inheritance, the mothers of patients 1 and 2 were balanced translocation carriers. Detecting cryptic balanced translocations is extremely important due to the high risk of recurrence, and enables the entire family to receive the appropriate genetic counselling. Patient 5 inherited the deletion from the father, who had a lower degree of ID than the proband; this suggests variable expressivity of the alteration. Patient 20 had a duplication on chromosome X which was inherited from the mother, who had no reported phenotype. This type of duplication involves genes associated with the disease according to the OMIM database. The fact that the mother did not exhibit ID/GDD may be due to a phenomenon of selective X inactivation. Lastly, the duplications in patients 19 and 22 were inherited from healthy mothers, and involved autosomes. Patient 19 showed a microduplication on chromosome 22; this variation has been associated with incomplete penetrance and highly variable expressivity, ranging from carriers with normal intelligence to carriers with severe ID and a large number of patients inheriting the alteration from an unaffected parent.18 Patient 22 had a duplication in a region frequently affected by microduplications/deletions on chromosome 15. The literature reports numerous cases of duplications inherited from a parent with normal phenotype. It has been hypothesised that manifestations are more severe when the variation is inherited from the mother, with great intrafamilial variability (from normal intelligence to severe cognitive impairment associated with epilepsy).19
In our sample, uncertain CNVs were inherited from a healthy parent in 90% of cases. This finding should be interpreted with caution: although there is still no conclusive evidence, these chromosomal imbalances may contribute to patients’ phenotypes as a result of variable expressivity of these variations, epigenetic factors, or recessive mutations in the non-deleted allele.14
Regarding anthropometric characteristics, previous studies have detected an association between microcephaly and pathological array CGH results.20 The percentage of patients with severe microcephaly (head circumference below −3 SD) was considerably higher in the group of patients with pathological array CGH results, although the difference was not statistically significant (Table 1). Based on the above, we hypothesise that we may have achieved similar results to those of the study cited above if we had included a larger sample. Other authors report higher frequency of growth disorders in patients with pathological array CGH results (slow growth according to Shoukier et al.20 and tall height in the study by Roselló et al.8); however, Vulto-van Silfhout et al.,15 in line with our results, found no association between growth and genetic test results. Contradictory evidence of the relationship between growth disorders and pathological array CGH results may be due to differences in the inclusion criteria established for each study and the results of array CGH (some syndromes are associated with slow growth, whereas others present with overgrowth). Further studies including larger sample sizes are necessary to determine the role of growth disorders in structural chromosomal alterations.
Numerous researchers have reported an association between chromosomal alterations and congenital malformations.8,15,20 In most series, the most prevalent congenital malformations were cardiac malformations; in our study, however, central nervous system malformations, especially those affecting the corpus callosum, were the most frequently observed. This finding coincides with the results of a study by Preiksaitiene et al.21 A review of the methodology of these studies shows that inclusion criteria determine, to a certain extent, the type of malformation which is most frequent in each sample. In our study, however, the distribution of malformations was entirely random.
The presence of dysmorphic craniofacial features was an extremely important variable in our sample due to the high prevalence of these anomalies in patients with ID/GDD. In fact, according to a thorough analysis of the presence of dysmorphic features, only 25% of the patients with normal array CGH results and 13.6% of those with pathological CNVs displayed no minor morphological alterations. In our study, significant inter-group differences were found in presence of unusual hair patterns, long philtrum, thin upper lip, and micrognathia. Although the most frequent dysmorphic feature in these patients varies greatly across series, the co-presence of several features in an individual constitutes a reliable indicator of possible chromosomal imbalances.15
Several studies have compared the diagnostic yields of different genetic tests for ID. Miller et al.13 reviewed 33 studies including a total of 21698 patients, concluding that the diagnostic yield of array CGH was higher (12% in unselected patients) than that of high-resolution karyotyping (3% excluding Down syndrome) and FISH assays for subtelomeric rearrangements (2.4%). A more recent study conducted in a European population of 36325 patients undergoing karyotyping and array CGH detected pathological CNVs in 19% of unselected patients; only 0.78% of these genetic alterations would have gone undetected without karyotyping.22 Both studies conclude that array CGH should be the first diagnostic tool in patients with idiopathic ID.
Due to the recent introduction of array CGH in clinical practice, there are still many limitations vis-à-vis correctly interpreting results and informing families. Previous studies have detected great variability in the interpretation of results and in the clinical management of these patients by different healthcare professionals.23 Both factors may improve if physicians and geneticists cooperate and design protocols aimed at reducing differences in management and increasing healthcare quality. Furthermore, as the technique becomes increasingly widespread in clinical practice, results are shared in public databases; these data, added to information on patients’ phenotypic characteristics, may help define pathogenicity of each of the alterations detected.
Regarding the cost-effectiveness of array CGH, a recent study concludes that molecular karyotyping is less costly than conventional karyotyping plus subsequent testing (e.g. FISH assays to measure telomere length) and achieves a 15%-20% increase in diagnostic yield compared to traditional techniques.24
At present, array CGH constitutes the gold standard technique for the study of ID/GDD since it has a higher diagnostic yield than conventional genetic tests (karyotyping, FISH/MLPA for telomere length measurement, etc.). Although several factors associated with a greater likelihood of chromosomal rearrangements have been detected, they do not have sufficient discriminant power to identify patients suitable for array CGH. Therefore, our hospital has modified its study protocol for ID/GDD; array CGH is now offered to all patients with ID/GDD of unknown origin.
FundingThis study has received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Caballero Pérez V, López Pisón FJ, Miramar Gallart MD, González Álvarez A, García Jiménez MC, García Iñiguez JP, et al. Fenotipo en pacientes con discapacidad intelectual y array-CGH patológico. Neurología. 2017;32:568–578.
This study has not been presented at any of the SEN's Annual Meetings or at any other meetings or congresses.
articles

Neurología (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals