



Las ataxias espinocerebelosas (SCA) son un grupo de enfermedades neurodegenerativas genética, clínica y patológicamente heterogéneo, caracterizado por presentar una ataxia cerebelosa lentamente progresiva.
ObjetivoIdentificar las vías nerviosas neurofisiológicamente afectadas, correlacionar los hallazgos con el tamaño de la expansión CAG y determinar la contribución del estudio neurofisiológico al diagnóstico diferencial de los dos genotipos más prevalentes en España, SCA2 y SCA3.
MétodoHemos examinado 10 pacientes SCA2 y 12 SCA3 mediante electromiografía, electroneurografía motora y sensitiva, potenciales evocados multimodales, estimulación magnética transcraneal, reflejo de parpadeo y mandibular. En el análisis estadístico empleamos estudios de regresión lineal, coeficiente de correlación de Spearman y el test no paramétrico “U de Mann-Whitney”.
ResultadosDetectamos anomalías compatibles con una neuronopatía sensitiva con axonopatía periférica en la mayoría de pacientes SCA2 y en una minoría de SCA3; la vía somatosensorial central presentó abundantes anomalías en ambas poblaciones. Registramos importantes alteraciones tronco-encefálicas en SCA2; particularmente, el reflejo maseterino estuvo alterado en todos los pacientes SCA2, manteniéndose intacto en los SCA3. El estudio de la vía córtico-espinal demostró un mayor porcentaje de anomalías en ambas poblaciones que estudios previos.
ConclusionesSCA2 es un modelo electrofisiológico sugestivo de una neuronopatía sensitiva con axonopatía periférica y central. Los estudios de las vías tronco-encefálicas demuestran una mayor incidencia de alteraciones en los pacientes SCA2. En los pacientes SCA3 se observaron importantes alteraciones de la vía somatosensorial central con relativa normalidad del estudio electroneurográfico. El reflejo mandibular fue el test de mayor utilidad en el diagnóstico diferencial de ambos genotipos.
The spinocerebellar ataxias (SCA) are a group of genetic neurodegenerative diseases, clinically and pathologically heterogeneous, characterized by slowly progressive cerebellar ataxia.
ObjectiveTo identify the neural pathways affected neurophysiologically, correlate the findings with the size of CAG expansion and determine the contribution of neurophysiological studies in the differential diagnosis of the two most prevalent genotypes in Spain, SCA2 and SCA3.
MethodWe examined 10 SCA2 and 12 SCA3 patients by electromyography, electroneurography motor and sensory, multimodal evoked potentials, transcranial magnetic stimulation, blink reflex and masseter reflex. In the statistical analysis linear regression studies were performed, and the, Spearman correlation coefficient and nonparametric test U of Mann-Whitney calculated.
ResultsWe detected the presence of a predominantly sensory neuropathy in most SCA2 patients and in a minority of SCA3 patients; the central somatosensory pathway showed significant defects in both populations. We recorded a high incidence of brain-stem electrophysiological abnormalities in SCA2 patients; in particular, the masseter reflex was abnormal in all SCA2 patients, remaining intact in all SCA3 patients. The study of cortico-spinal pathway showed a greater percentage of abnormalities in both populations than in previous studies.
ConclusionSCA2 is a model of sensory neuronopathy with central and peripheral axonopathy. Studies of brain-stem pathways show a higher incidence of abnormalities in SCA2 patients. SCA3 patients show major changes in the central somatosensory pathway with relative normality of the electroneurography. The masseter reflex was the most useful test in the differential diagnosis between both genotypes.
Las ataxias cerebelosas con transmisión autosómica dominante son un grupo de enfermedades neurodegenerativas caracterizadas por ataxia cerebelosa lentamente progresiva como síntoma principal, causada por la degeneración del cerebelo y de sus conexiones aferentes y eferentes. En la mayoría de las familias se han detectado, además, evidencias clínicas y patológicas de afectación en otras estructuras del sistema nervioso tales como sistema extrapiramidal, oculomotor, sistema nervioso periférico y médula espinal1. Los recientes estudios de genética molecular han detectado que el defecto molecular más frecuente es una expansión dinámica del triplete CAG que codifica para tractos de poliglutamina2,3, habiéndose localizado 30 loci diferentes4 designados con el término “ataxia espinocerebelosa” (SCA1-30, acrónimo derivado de spinocerebellar ataxia en la literatura anglosajona).
La prevalencia de las SCA varía de unos países a otros, destacando en España un predominio de los genotipos SCA2 y SCA32,5. El objetivo principal de nuestro estudio fue identificar neurofisiológicamente las vías nerviosas alteradas, establecer las correlaciones de las alteraciones neurofisiológicas observadas con el tamaño de la expansión (CAG) subyacente y determinar la posible contribución del estudio neurofisiológico al diagnóstico diferencial de los genotipos SCA2 y SCA3.
Pacientes y métodoSe estudiaron aquellos pacientes con clínica de ataxia cerebelosa de inicio tardío cuyo estudio genético había demostrado la existencia de la mutación subyacente para SCA2 y SCA3. Asimismo, se estudiaron los miembros pertenecientes a la familia del “propósitus” que presentaban la mutación, ya fueran portadores asintomáticos o sintomáticos. Para detalles del estudio molecular remitimos al lector a la referencia de Infante et al5. El estudio fue aprobado por el Comité de Ética de Cantabria.
En definitiva, se estudiaron 10 pacientes (7 mujeres y 3 varones) de 6 familias SCA2 no relacionadas entre sí, con una edad media en el momento del estudio de 48,3±12,7 años y una duración media de la enfermedad de 9,8±4,3 años, y 12 miembros (3 mujeres y 9 varones) de tres familias SCA3, no relacionadas entre sí, con una edad media de 43,6±11,7 años y una duración media de la enfermedad de 7,3±5,8 años en el momento del estudio.
No se encontraron diferencias estadísticamente significativas entre ambas poblaciones para la edad media de los pacientes ni para la duración media de la enfermedad (p>0,05).
ElectromiografíaSe llevó a cabo con electrodos de aguja concéntricos que se insertaron en el vientre muscular del músculo tibialis anterior (TA), registrando potenciales de unidad motora, de los que analizamos duración y morfología. Además se registró también la actividad muscular espontánea y el patrón de máximo esfuerzo. Únicamente en dos de los pacientes SCA2 con mioquimias clínicas que realizó registro electromiográfico de la musculatura facial.
ElectroneurografíaSe estudiaron los nervios mediano y peroneal mediante la estimulación en diferentes puntos de su trayecto con electrodos de superficie bipolar, registrando los potenciales motores con electrodos de superficie situando el activo sobre el vientre muscular y la referencia sobre la inserción tendinosa. Analizamos la amplitud del potencial motor, la latencia motora distal (LMD), la velocidad de conducción motora (VCM) y la latencia mínima de las ondas F. Tomamos como valores de normalidad los presentados por Preston y Shapiro6.
Se utilizó el método de registro ortodrómico para estudiar el componente sensitivo del nervio mediano, según el cual el potencial sensitivo fue registrado proximalmente al estímulo eléctrico. El estimulador que empleamos estaba formado por dos electrodos de anillo flexibles que se adaptan al perímetro de los dedos (primero y tercero) situando el ánodo en la falange distal y el cátodo 2cm proximalmente. Únicamente empleamos el método de registro antidrómico al explorar el nervio sural, de este modo el potencial sensitivo fue registrado detrás del maléolo externo, ante un estímulo eléctrico (cátodo distal) que aplicamos 14cm proximal al electrodo de registro. El potencial sensitivo en ambos nervios se registró con electrodos de superficie y los parámetros estudiados fueron la amplitud pico-pico del potencial sensitivo y la velocidad de conducción sensitiva (VCS).
Para los potenciales sensitivos de nervio mediano tomamos los valores de normalidad obtenidos en nuestro laboratorio, mientras que para el nervio sural empleamos los presentados por Preston y Shapiro6.
Potenciales evocados somatosensorialesSe obtuvieron estimulando el nervio mediano en la muñeca situando los electrodos de registro en el área postrolándica contralateral (posiciones C3’/C4’) y en la región cervical (Cv7) con referencia en la región frontal (Fpz). Los potenciales evocados somatosensoriales (PESS) de nervio tibial posterior se obtuvieron estimulando este nervio detrás del maléolo interno, registrando la respuesta 2cm posterior al vértex cerebral (posición Cz’), también con referencia cefálica (Fpz). Analizamos la latencia y amplitud de los distintos componentes, así como el tiempo de conducción somatosensorial central (TCSC).
Empleamos como valores de normalidad los presentados por Chiappa7.
Estimulación magnéticaLos potenciales evocados motores (PEM) se obtuvieron mediante estimulación magnética transcraneal (EMT), realizada con un equipo Magstim (Modelo 200 con Hight Power 90mm Coil), sobre el córtex motor, la región cervical y lumbar con registro mediante electrodos de superficie situados en los músculos abductor pollicis brevis (APB) en extremidades superiores y extensor digitorum brevis (EDB) o TA en extremidades inferiores, en situación de reposo. Analizamos la latencia absoluta del PEM y el tiempo de conducción motora central (TCMC) por el método de la onda F descrito por Rossini et al8.
Empleamos la técnica y los valores de normalidad obtenidos en nuestro laboratorio y presentados por Calleja et al9.
Potenciales evocados visualesSe registraron en las áreas occipitales (O1, Oz y O2) con referencia en Fz estimulando ambos ojos de forma independiente con una pantalla tipo damero (PEV-pattern). Analizamos latencia y amplitud de la onda P100.
Hemos tomado como referencia los valores de normalidad presentados por Chiappa7.
Potenciales evocados auditivosSe obtuvieron aplicando un estímulo acústico a 80 dBnHL de intensidad con registro de la respuesta en la derivación lóbulo de la oreja ipsilateral (A1/ A2)- vértex (Cz). Analizamos las latencias absolutas e interpicos de los distintos componentes de la respuesta.
Hemos tomado como referencia los valores de normalidad presentados por Chiappa7.
Reflejo de parpadeoSe obtuvo estimulando el nervio supraorbitario a su salida por el orificio frontal lateral. El registro se realizó simultáneamente, mediante electrodos de superficie situados en ambos músculos orbicularis oculi, analizando las latencias de las respuestas R1 y R2 ipsilateral al estímulo y la respuesta R2 contralateral al mismo.
Los valores de normalidad se tomaron de los presentados por Kimura et al10.
Reflejo mandibularPara la obtención del reflejo mandibular empleamos un martillo de reflejos conectado al amplificador, mediante el cual aplicamos un estímulo mecánico sobre el mentón interponiendo un dedo del explorador. El registro se efectuó mediante electrodos de superficie situados sobre el músculo masseter (activo: vientre muscular; referencia: 2cm por debajo del ángulo de la mandíbula) en ambos lados. Así obtuvimos de manera bilateral y simultánea al menos 10 respuestas consecutivas. Posteriormente, se valoró la existencia o no de respuesta refleja y la latencia de la misma.
Se tomaron como referencia los valores de normalidad presentados por Cruccu et al11.
Análisis estadísticoEmpleamos el paquete estadístico SPSS 8.0 para el cálculo de medias, desviación estándar y rango de todos los parámetros electrofisiológicos obtenidos. El estudio de regresión lineal y el coeficiente de correlación de Spearman nos permitieron determinar si existe algún tipo de relación (grado y dirección) entre los distintos parámetros electrofisiológicos obtenidos y el número de repeticiones CAG de la mutación subyacente, y para la comparación de medias entre ambas poblaciones (SCA2 vs SCA3) empleamos el test no paramétrico “U de Mann-Whitney” para muestras pequeñas. En la comparación de proporciones empleamos la prueba de Chi cuadrado aplicando el test exacto de Fisher. La significación estadística se estableció para un valor de p<0,05.
ResultadosEl registro electromiográfico del músculo TA en los pacientes de SCA2 evidenció parámetros normales excepto en 2 (20%) casos en los que se registró un patrón neurógeno crónico. Asimismo, sólo 2 (16%) pacientes de SCA3 mostraron el mismo patrón en el registro electromiográfico del músculo TA.
Clínicamente se observaron mioquimias faciales en 6 (67%) pacientes de SCA2 y en un (8%) paciente de SCA3. El registro EMG de la musculatura facial evidenció descargas repetitivas espontáneas de potenciales de unidad motora aislados y agrupados con una frecuencia de descarga de 22Hz compatibles con mioquimias en dos pacientes de SCA2.
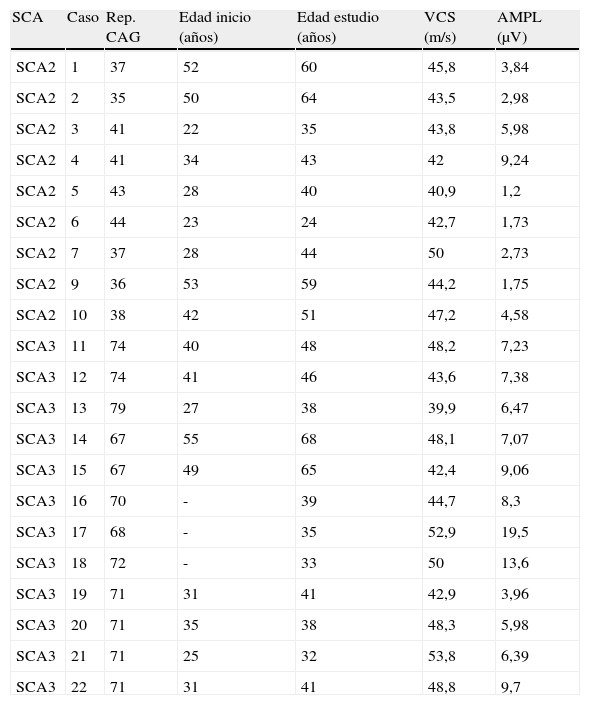
La electroneurografía presentó las anomalías más relevantes en el estudio del nervio sural, destacando una reducción en la amplitud del potencial sensitivo en 8 (88%) de los 9 pacientes de SCA2 estudiados frente a sólo 2 (16%) de los 12 pacientes de SCA3, con normalidad en la VCS (tabla 1). El análisis estadístico únicamente reveló que la amplitud de los potenciales sensitivos tanto de nervio mediano como sural eran significativamente menores en los pacientes de SCA2 (p<0,01). No se encontró ninguna relación entre los parámetros obtenidos y el número de repeticiones CAG de la mutación.
Nervio sural
| SCA | Caso | Rep. CAG | Edad inicio (años) | Edad estudio (años) | VCS (m/s) | AMPL (μV) |
| SCA2 | 1 | 37 | 52 | 60 | 45,8 | 3,84 |
| SCA2 | 2 | 35 | 50 | 64 | 43,5 | 2,98 |
| SCA2 | 3 | 41 | 22 | 35 | 43,8 | 5,98 |
| SCA2 | 4 | 41 | 34 | 43 | 42 | 9,24 |
| SCA2 | 5 | 43 | 28 | 40 | 40,9 | 1,2 |
| SCA2 | 6 | 44 | 23 | 24 | 42,7 | 1,73 |
| SCA2 | 7 | 37 | 28 | 44 | 50 | 2,73 |
| SCA2 | 9 | 36 | 53 | 59 | 44,2 | 1,75 |
| SCA2 | 10 | 38 | 42 | 51 | 47,2 | 4,58 |
| SCA3 | 11 | 74 | 40 | 48 | 48,2 | 7,23 |
| SCA3 | 12 | 74 | 41 | 46 | 43,6 | 7,38 |
| SCA3 | 13 | 79 | 27 | 38 | 39,9 | 6,47 |
| SCA3 | 14 | 67 | 55 | 68 | 48,1 | 7,07 |
| SCA3 | 15 | 67 | 49 | 65 | 42,4 | 9,06 |
| SCA3 | 16 | 70 | - | 39 | 44,7 | 8,3 |
| SCA3 | 17 | 68 | - | 35 | 52,9 | 19,5 |
| SCA3 | 18 | 72 | - | 33 | 50 | 13,6 |
| SCA3 | 19 | 71 | 31 | 41 | 42,9 | 3,96 |
| SCA3 | 20 | 71 | 35 | 38 | 48,3 | 5,98 |
| SCA3 | 21 | 71 | 25 | 32 | 53,8 | 6,39 |
| SCA3 | 22 | 71 | 31 | 41 | 48,8 | 9,7 |
VCS: velocidad de conducción sensitiva; AMPL: amplitud; -: asintomático.
Los PESS de nervio mediano presentaron anomalías en 7 (77%) pacientes de SCA2 y en 5 (42%) pacientes de SCA3, entre las que destaca la ausencia de alguno de los componentes de la respuesta (N20 y /o N13), el retraso de sus latencias y el incremento en el TCSC (N13-N20). Las anomalías en los PESS de nervio tibial posterior se detectaron en 8 (88%) pacientes de SCA2 y 8 (66%) pacientes de SCA3, destacando la ausencia de respuesta cortical P40, el retraso de la misma y/o la reducción en su amplitud. Todos los pacientes con anomalías en los PESS procedentes de extremidades superiores presentaron también anomalías en la respuesta procedente de extremidades inferiores. El análisis comparativo demostró que únicamente el TCSC se encontraba significativamente más prolongado en los pacientes de SCA2 (p<0,05), no encontrando diferencias significativas para el resto de parámetros. Tampoco se encontró ninguna relación entre los parámetros obtenidos y el tamaño de la expansión.
El estudio de la vía córtico-espinal puso de manifiesto anomalías en 5 (55%) pacientes de SCA2 y en 3 (25%) pacientes de SCA3, que consistieron en la ausencia del PEM en extremidades inferiores y en incrementos del TCMC tanto en miembros superiores como inferiores. No se encontraron diferencias significativas para el TCMC entre ambas poblaciones, así como ninguna relación entre estos parámetros y el tamaño de la expansión.
En los pacientes de SCA2 estudiados, 5 (71%) presentaron anomalías en el estudio de la vía visual; en tres de ellos la amplitud de la respuesta P100 estaba reducida, en uno la latencia se hallaba prolongada y en el caso restante se detectaron ambas anomalías. En los pacientes de SCA3 sólo 2 (16%) presentaron una amplitud reducida para la onda P100. A pesar de ello, el análisis comparativo no demostró diferencias significativas para las dos variables (amplitud y latencia) estudiadas. El análisis estadístico tampoco demostró una relación significativa entre estas variables y el tamaño de la expansión subyacente.
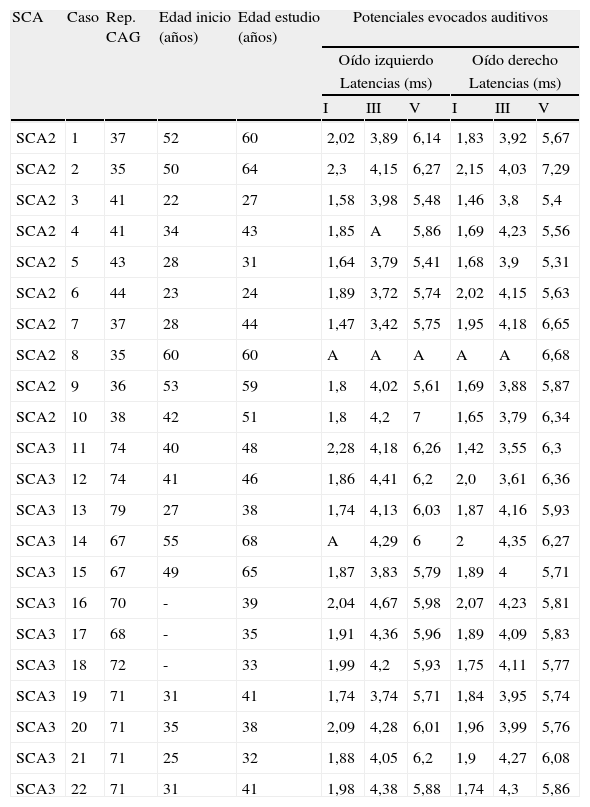
Los potenciales evocados auditivos (PEAT) presentaron alteraciones en 4 (40%) pacientes de SCA2 y en 2 (17%) pacientes de SCA3, entre las que destacan un retraso de las latencias absolutas y/o interpicos de alguno de los componentes de la respuesta junto a una mala replicabilidad de la misma (tabla 2). Sin embargo, no se hallaron diferencias significativas entre ambas poblaciones, ni una relación significativa entre estas variables y el tamaño de la mutación.
Potenciales evocados auditivos
| SCA | Caso | Rep. CAG | Edad inicio (años) | Edad estudio (años) | Potenciales evocados auditivos | |||||
| Oído izquierdo | Oído derecho | |||||||||
| Latencias (ms) | Latencias (ms) | |||||||||
| I | III | V | I | III | V | |||||
| SCA2 | 1 | 37 | 52 | 60 | 2,02 | 3,89 | 6,14 | 1,83 | 3,92 | 5,67 |
| SCA2 | 2 | 35 | 50 | 64 | 2,3 | 4,15 | 6,27 | 2,15 | 4,03 | 7,29 |
| SCA2 | 3 | 41 | 22 | 27 | 1,58 | 3,98 | 5,48 | 1,46 | 3,8 | 5,4 |
| SCA2 | 4 | 41 | 34 | 43 | 1,85 | A | 5,86 | 1,69 | 4,23 | 5,56 |
| SCA2 | 5 | 43 | 28 | 31 | 1,64 | 3,79 | 5,41 | 1,68 | 3,9 | 5,31 |
| SCA2 | 6 | 44 | 23 | 24 | 1,89 | 3,72 | 5,74 | 2,02 | 4,15 | 5,63 |
| SCA2 | 7 | 37 | 28 | 44 | 1,47 | 3,42 | 5,75 | 1,95 | 4,18 | 6,65 |
| SCA2 | 8 | 35 | 60 | 60 | A | A | A | A | A | 6,68 |
| SCA2 | 9 | 36 | 53 | 59 | 1,8 | 4,02 | 5,61 | 1,69 | 3,88 | 5,87 |
| SCA2 | 10 | 38 | 42 | 51 | 1,8 | 4,2 | 7 | 1,65 | 3,79 | 6,34 |
| SCA3 | 11 | 74 | 40 | 48 | 2,28 | 4,18 | 6,26 | 1,42 | 3,55 | 6,3 |
| SCA3 | 12 | 74 | 41 | 46 | 1,86 | 4,41 | 6,2 | 2,0 | 3,61 | 6,36 |
| SCA3 | 13 | 79 | 27 | 38 | 1,74 | 4,13 | 6,03 | 1,87 | 4,16 | 5,93 |
| SCA3 | 14 | 67 | 55 | 68 | A | 4,29 | 6 | 2 | 4,35 | 6,27 |
| SCA3 | 15 | 67 | 49 | 65 | 1,87 | 3,83 | 5,79 | 1,89 | 4 | 5,71 |
| SCA3 | 16 | 70 | - | 39 | 2,04 | 4,67 | 5,98 | 2,07 | 4,23 | 5,81 |
| SCA3 | 17 | 68 | - | 35 | 1,91 | 4,36 | 5,96 | 1,89 | 4,09 | 5,83 |
| SCA3 | 18 | 72 | - | 33 | 1,99 | 4,2 | 5,93 | 1,75 | 4,11 | 5,77 |
| SCA3 | 19 | 71 | 31 | 41 | 1,74 | 3,74 | 5,71 | 1,84 | 3,95 | 5,74 |
| SCA3 | 20 | 71 | 35 | 38 | 2,09 | 4,28 | 6,01 | 1,96 | 3,99 | 5,76 |
| SCA3 | 21 | 71 | 25 | 32 | 1,88 | 4,05 | 6,2 | 1,9 | 4,27 | 6,08 |
| SCA3 | 22 | 71 | 31 | 41 | 1,98 | 4,38 | 5,88 | 1,74 | 4,3 | 5,86 |
A: ausencia de respuesta; -: asintomático.
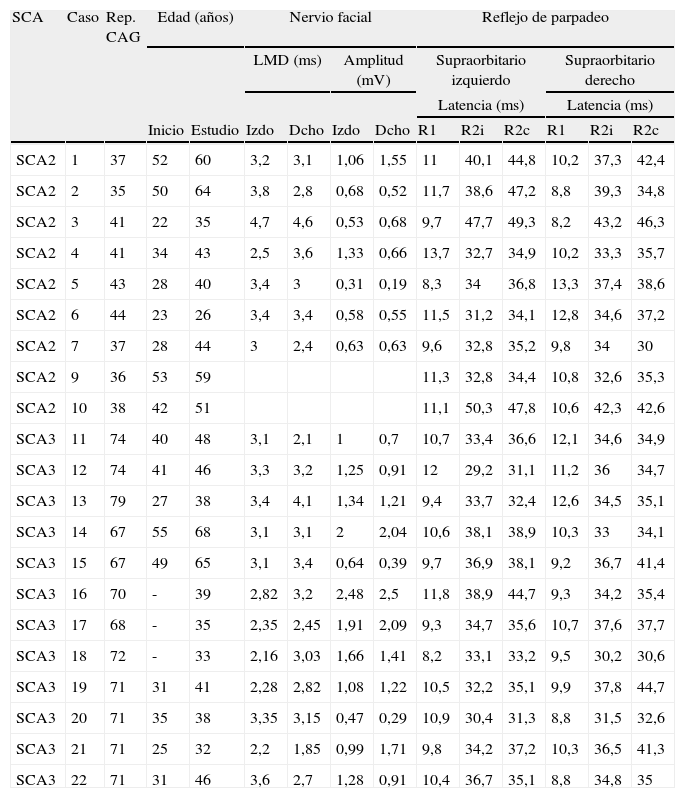
En el estudio del reflejo de parpadeo se detectaron anomalías en 6 (67%) de los 9 pacientes con la mutación SCA2 estudiados; en 4 de ellos las anomalías se correspondían con un retraso en la latencia de los componentes polisinápticos de la respuesta y en los otros dos el retraso se detectó en la respuesta directa R1. En los pacientes SCA3 sólo se apreció un retraso en la respuesta polisináptica R2 contralateral en 2 (17%) pacientes (tabla 3). Sin embargo, no se encontraron diferencias significativas entre ambas poblaciones para las latencias de los distintos componentes del arco reflejo.
Nervio facial y reflejo de parpadeo
| SCA | Caso | Rep. CAG | Edad (años) | Nervio facial | Reflejo de parpadeo | |||||||||
| LMD (ms) | Amplitud (mV) | Supraorbitario izquierdo | Supraorbitario derecho | |||||||||||
| Latencia (ms) | Latencia (ms) | |||||||||||||
| Inicio | Estudio | Izdo | Dcho | Izdo | Dcho | R1 | R2i | R2c | R1 | R2i | R2c | |||
| SCA2 | 1 | 37 | 52 | 60 | 3,2 | 3,1 | 1,06 | 1,55 | 11 | 40,1 | 44,8 | 10,2 | 37,3 | 42,4 |
| SCA2 | 2 | 35 | 50 | 64 | 3,8 | 2,8 | 0,68 | 0,52 | 11,7 | 38,6 | 47,2 | 8,8 | 39,3 | 34,8 |
| SCA2 | 3 | 41 | 22 | 35 | 4,7 | 4,6 | 0,53 | 0,68 | 9,7 | 47,7 | 49,3 | 8,2 | 43,2 | 46,3 |
| SCA2 | 4 | 41 | 34 | 43 | 2,5 | 3,6 | 1,33 | 0,66 | 13,7 | 32,7 | 34,9 | 10,2 | 33,3 | 35,7 |
| SCA2 | 5 | 43 | 28 | 40 | 3,4 | 3 | 0,31 | 0,19 | 8,3 | 34 | 36,8 | 13,3 | 37,4 | 38,6 |
| SCA2 | 6 | 44 | 23 | 26 | 3,4 | 3,4 | 0,58 | 0,55 | 11,5 | 31,2 | 34,1 | 12,8 | 34,6 | 37,2 |
| SCA2 | 7 | 37 | 28 | 44 | 3 | 2,4 | 0,63 | 0,63 | 9,6 | 32,8 | 35,2 | 9,8 | 34 | 30 |
| SCA2 | 9 | 36 | 53 | 59 | 11,3 | 32,8 | 34,4 | 10,8 | 32,6 | 35,3 | ||||
| SCA2 | 10 | 38 | 42 | 51 | 11,1 | 50,3 | 47,8 | 10,6 | 42,3 | 42,6 | ||||
| SCA3 | 11 | 74 | 40 | 48 | 3,1 | 2,1 | 1 | 0,7 | 10,7 | 33,4 | 36,6 | 12,1 | 34,6 | 34,9 |
| SCA3 | 12 | 74 | 41 | 46 | 3,3 | 3,2 | 1,25 | 0,91 | 12 | 29,2 | 31,1 | 11,2 | 36 | 34,7 |
| SCA3 | 13 | 79 | 27 | 38 | 3,4 | 4,1 | 1,34 | 1,21 | 9,4 | 33,7 | 32,4 | 12,6 | 34,5 | 35,1 |
| SCA3 | 14 | 67 | 55 | 68 | 3,1 | 3,1 | 2 | 2,04 | 10,6 | 38,1 | 38,9 | 10,3 | 33 | 34,1 |
| SCA3 | 15 | 67 | 49 | 65 | 3,1 | 3,4 | 0,64 | 0,39 | 9,7 | 36,9 | 38,1 | 9,2 | 36,7 | 41,4 |
| SCA3 | 16 | 70 | - | 39 | 2,82 | 3,2 | 2,48 | 2,5 | 11,8 | 38,9 | 44,7 | 9,3 | 34,2 | 35,4 |
| SCA3 | 17 | 68 | - | 35 | 2,35 | 2,45 | 1,91 | 2,09 | 9,3 | 34,7 | 35,6 | 10,7 | 37,6 | 37,7 |
| SCA3 | 18 | 72 | - | 33 | 2,16 | 3,03 | 1,66 | 1,41 | 8,2 | 33,1 | 33,2 | 9,5 | 30,2 | 30,6 |
| SCA3 | 19 | 71 | 31 | 41 | 2,28 | 2,82 | 1,08 | 1,22 | 10,5 | 32,2 | 35,1 | 9,9 | 37,8 | 44,7 |
| SCA3 | 20 | 71 | 35 | 38 | 3,35 | 3,15 | 0,47 | 0,29 | 10,9 | 30,4 | 31,3 | 8,8 | 31,5 | 32,6 |
| SCA3 | 21 | 71 | 25 | 32 | 2,2 | 1,85 | 0,99 | 1,71 | 9,8 | 34,2 | 37,2 | 10,3 | 36,5 | 41,3 |
| SCA3 | 22 | 71 | 31 | 46 | 3,6 | 2,7 | 1,28 | 0,91 | 10,4 | 36,7 | 35,1 | 8,8 | 34,8 | 35 |
Dcho: derecho; Izdo: izquierdo; R2i: R2 ipsilateral; R2c: R2 contralateral; -: asintomático.
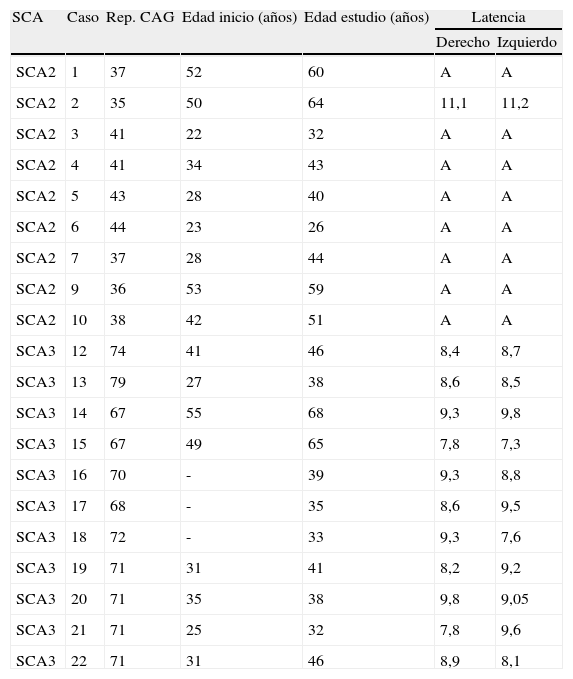
Finalmente, el reflejo mandibular fue patológico en los 9 (100%) pacientes de SCA2 estudiados; no se obtuvo reflejo en 8 de ellos y en el restante se esbozaba una respuesta de amplitud reducida (< 0,1mV) y latencia prolongada bilateralmente (11,1 y 11,2ms). Por el contrario, los 12 (100%) pacientes de SCA3 explorados presentaron un reflejo mandibular normal (latencia < 10ms) (tabla 4). El análisis comparativo de proporciones entre ambas poblaciones SCA2 y SCA3 mostró una diferencia significativa (p<0,001).
Reflejo mandibular
| SCA | Caso | Rep. CAG | Edad inicio (años) | Edad estudio (años) | Latencia | |
| Derecho | Izquierdo | |||||
| SCA2 | 1 | 37 | 52 | 60 | A | A |
| SCA2 | 2 | 35 | 50 | 64 | 11,1 | 11,2 |
| SCA2 | 3 | 41 | 22 | 32 | A | A |
| SCA2 | 4 | 41 | 34 | 43 | A | A |
| SCA2 | 5 | 43 | 28 | 40 | A | A |
| SCA2 | 6 | 44 | 23 | 26 | A | A |
| SCA2 | 7 | 37 | 28 | 44 | A | A |
| SCA2 | 9 | 36 | 53 | 59 | A | A |
| SCA2 | 10 | 38 | 42 | 51 | A | A |
| SCA3 | 12 | 74 | 41 | 46 | 8,4 | 8,7 |
| SCA3 | 13 | 79 | 27 | 38 | 8,6 | 8,5 |
| SCA3 | 14 | 67 | 55 | 68 | 9,3 | 9,8 |
| SCA3 | 15 | 67 | 49 | 65 | 7,8 | 7,3 |
| SCA3 | 16 | 70 | - | 39 | 9,3 | 8,8 |
| SCA3 | 17 | 68 | - | 35 | 8,6 | 9,5 |
| SCA3 | 18 | 72 | - | 33 | 9,3 | 7,6 |
| SCA3 | 19 | 71 | 31 | 41 | 8,2 | 9,2 |
| SCA3 | 20 | 71 | 35 | 38 | 9,8 | 9,05 |
| SCA3 | 21 | 71 | 25 | 32 | 7,8 | 9,6 |
| SCA3 | 22 | 71 | 31 | 46 | 8,9 | 8,1 |
A: ausencia de respuesta; -: asintomático.
En la mayoría de los pacientes de SCA2 apreciamos signos electrofisiológicos compatibles con una neuronopatía sensitiva con axonopatía periférica. Estos resultados coinciden en gran medida con las anomalías del sistema nervioso periférico descritas en pacientes con atrofia olivopontocerebelosa12–16 y en pacientes genéticamente tipificados como SCA217–20. También están en concordancia con los hallazgos histológicos detectados en las necropsias de pacientes con dichas neurodegeneraciones, como son la pérdida de neuronas de los ganglios de la raíz dorsal, con la consiguiente degeneración de las fibras mielínicas de grueso y mediano calibre de las raíces dorsales y de las columnas posteriores de la médula espinal14,16,17,21,22. En nuestros pacientes de SCA3 apenas hemos encontrado signos de neuropatía periférica, a diferencia de lo descrito por otros autores18–21,23–26. Creemos que estas discrepancias pueden deberse a la gran variabilidad fenotípica de esta enfermedad, pues sólo 2 de los 12 pacientes SCA3 podrían englobarse en el subtipo III, caracterizado por presentar signos prominentes de polineuropatía distal.
En ambos genotipos SCA2 y SCA3 encontramos anomalías en la vía somatosensorial similares a las descritas previamente por otros autores en pacientes de SCA218,19,27–29 y de SCA323,29. Estas alteraciones se han relacionado con la pérdida de fibras de las columnas dorsales de la médula espinal detectada en la autopsia de pacientes SCA230,31 y SCA332,33.
Nuestros resultados sugieren que la afectación de la vía córtico-espinal, tanto en pacientes SCA2 como SCA3 es más frecuente de lo descrito hasta este momento28,29,34,35. Creemos que esta discordancia puede deberse a la evaluación de la vía en extremidades superiores e inferiores, puesto que la severidad del proceso patológico parece depender de la longitud de los tractos motores28. Otro factor a tener en cuenta es la duración de la enfermedad pues sabemos que la afectación de la vía córtico-espinal depende del momento evolutivo del proceso degenerativo36,37. De hecho, en nuestro estudio, hemos observado que aquellos pacientes con anomalías en la vía córtico-espinal fueron los que presentaban un cuadro clínico de mayor duración (hasta 16 años).
Un importante número pacientes de SCA2 mostraron anomalías en los PEV que coinciden con los hallazgos descritos por autores como Perreti et al28 y discrepan con otros autores como Abele et al29 y Velázquez et al18, cuyos estudios demostraron una tendencia a la conservación de la vía visual de estos pacientes. En esta variabilidad pueden influir las diferencias metodológicas de la técnica empleada para la obtención de los PEV. Aunque no hay estudios neuropatológicos de la vía visual en estos pacientes, Abele et al29 sugieren que las alteraciones del PEV pueden estar relacionadas con una pérdida de fibras mielínicas de grueso y mediano calibre en el nervio óptico. En SCA3 nuestros resultados sugieren una conservación de la vía visual coincidiendo con los resultados presentados previamente23,29.
Casi la mitad de nuestros pacientes de SCA2, frente a una minoría de pacientes de SCA3, mostraron alteraciones en los PEAT que sugieren la presencia de una afectación difusa de la vía auditiva, tanto del componente periférico como de la parte tronco-encefálica de la misma. Estos hallazgos son similares a los descritos por Perreti et al28 y Abele et al29. La alta incidencia de anomalías en los pacientes de SCA2 se ha asociado con la presencia de importantes alteraciones tronco-encefálicas en los estudios anatomopatológicos y de neuroimagen.
La totalidad de nuestros pacientes de SCA2 presentaron alteraciones en el reflejo mandibular38, y aunque la mayoría de estos pacientes presentaron signos sugestivos de neuronopatía, esta no puede ser la responsable de dichas alteraciones puesto que las fibras que constituyen la vía aferente del arco reflejo están situadas en el núcleo mesencefálico del trigémino (tronco cerebral) y no en los ganglios cráneo-espinales39. Así, la alteración patológica responsable podría encontrarse en el propio tronco cerebral, hecho que se puede apoyar en los hallazgos anatomopatológicos descritos en pacientes SCA2, en los que se han detectado una pérdida acusada de motoneuronas en todos los núcleos del trigémino21,31; contrariamente no se han descrito tales anomalías en los estudios anatomo-patológicos realizados a pacientes de SCA332,33,40,41, lo cual se correlaciona con la presencia del reflejo mandibular intacto en todos los pacientes de SCA3 de nuestro estudio.
Las anomalías halladas en el reflejo de parpadeo de la mayoría de nuestros pacientes de SCA2 tampoco se pueden relacionar con una neuronopatía, puesto que las fibras que constituyen la vía aferente, aunque se encuentran en el ganglio de Gasser, son fibras de medio y pequeño calibre que no se ven afectas en este proceso. Ello nos induce a pensar que la causa responsable de estas anomalías se encuentra localizada en el tronco cerebral, concretamente en los núcleos principal y espinal del trigémino, que como ya hemos visto se hallan severamente dañados en los pacientes de SCA231.
Finalmente, el registro electromiográfico corroboró la presencia clínica de mioquimias faciales en los pacientes de SCA2. Según los estudios de Valls-Solé et al42 podemos sugerir que la alta incidencia de mioquimias faciales se debe a una hiperexcitabilidad neuronal tronco-encefálica, mientras que la baja incidencia de las mismas junto con la escasa afectación de los reflejos tronco-encefálicos en pacientes de SCA3 estaría acorde con las escasas alteraciones de las estructuras tronco-encefálicas que presentan estos pacientes.
ConclusionesLa vía somatosensorial y el componente sensitivo del nervio periférico fueron los sistemas que con más frecuencia se encontraron afectados en los pacientes de SCA2, corroborando que SCA2 es compatible con una neuronopatía sensitiva con axonopatía central y periférica. A este hecho podemos añadir la elevada incidencia de alteraciones detectadas en el estudio de las vías tronco-encefálicas, que están en consonancia con las importantes anomalías apreciadas en estas estructuras en los estudios anatomo-patológicos y de neuroimagen en este genotipo.
En los pacientes de SCA3 detectamos alteraciones en la vía somatosensorial en dos tercios de los pacientes con conservación del sistema nervioso periférico. Esta discrepancia respecto a estudios previos podría deberse a la gran variabilidad fenotípica de la mutación SCA3; de hecho en nuestra serie sólo dos casos pertenecían al subtipo III. Del mismo modo que el sistema nervioso periférico, el estudio de las vías nerviosas tronco-encefálicas (PEAT, reflejo mandibular y de parpadeo) fue normal en la mayoría de los pacientes SCA3, de acuerdo con la relativa preservación del tallo cerebral en este genotipo.
Nuestros resultados sugieren que la afectación de la vía córtico-espinal es más frecuente de lo reportado por otros autores en ambos genotipos, probablemente porque para su demostración es preciso evaluar las vías motoras centrales de miembros superiores e inferiores.
Comparando los resultados en SCA2 y SCA3 no hemos encontrado diferencias significativas en los estudios de las vías somatosensorial y córtico-espinal; los estudios de la vía auditiva y del reflejo de parpadeo mostraron un mayor porcentaje de alteraciones en SCA2, pero la diferencia tampoco fue significativa.
Finalmente, la alteración del reflejo mandibular en todos los pacientes SCA2 frente a su conservación en todos los SCA3 hace que esta sencilla exploración pueda ser de gran utilidad en el diagnóstico diferencial de ambos genotipos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
AgradecimientosA la Srta. Rosario Repoila, por la asistencia técnica.
Este trabajo ha sido patrocinado por el Fondo de Investigación Sanitaria (FIS PI07/1323E) y EUROSCA.

