










a través de material exógeno que contenga priones, y han recibido el nombre de enfermedad de Creutzfeldt-Jakob (ECJ), enfermedad de Gerstman-Straüssler-Scheinken (GSS), insomnio familiar letal (IFL) y kuru. Neuropatológicamente se caracterizan por la presencia de degeneración espongiforme, astrocitosis y pérdida neuronal, en ausencia de infiltrados inflamatorios. En este artículo se revisan las principales características de las ETT que afectan a humanos y a animales.
Las principales encefalopatías espongiformes transmisibles (EET) se enumeran en la tabla 1, tanto las que afectan a humanos como las que lo hacen a diversos tipos de animales1-4.

Biología de las encefalopatías espongiformes transmisibles
El scrapie de ovejas y cabras, prototipo de enfermedad priónica, se describió por primera vez en 1732, y es la primera EET conocida. En 1936, por inoculación experimental, se demuestra la transmisibilidad del scrapie de oveja a oveja5, y posteriormente, en los años sesenta, la del kuru y ECJ a primates6.
Naturaleza del prión
Fue Prusiner en 1982 quien introdujo el término "prión" (proteinaceous infective particle) para designar a una isoforma anómala, PrPsc o PrPres, de una proteína celular normal, la proteína priónica (PrPc), con capacidad autopropagativa e infectiva y que era resistente a proteasas, insoluble en detergentes y con capacidad de recuperar su antigenicidad tras la desnaturalización3,4.
La proteína priónica (PrPc) se codifica en el ser humano por un gen situado en el brazo corto del cromosoma 20, y tiene un peso molecular de 33-35 kD. Se sabe que la PrPc se encuentra asociada a la membrana plasmática a través de un residuo de fosfatidilinositol glucosilado, pero se desconoce su función exacta.
La proteína priónica normal y la patológica comparten la misma estructura primaria, esto es, la secuencia de aminoácidos y la diferencia entre ambas isoformas es su conformación. La PrPc se pliega en hélices-* principalmente, y posee escasas láminas-ß. La conversión de PrPc a PrPsc implica el desplegamiento de hélices-* y su replegamiento en láminas-ß (fig. 1). Este cambio conformacional es lo que le confiere a la proteína priónica capacidad autorreplicativa, además de resistencia a diversos agentes fisicoquímicos. Según el patrón de glucosilación y el fragmento resistente a proteasas se definen las distintas cepas de PrPsc; hasta el momento se han caracterizado 5 tipos de PrPsc.
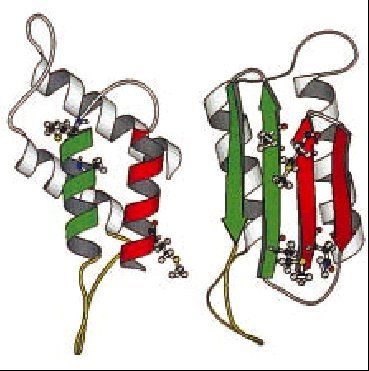
Fig. 1. Proteína priónica normal y patológica.
Patogenia
La transmisión de la enfermedad se ha demostrado por vía oral, por inyección intramuscular o intraperitoneal, por instrumental quirúrgico, por implantes corneales o de duramadre contaminados y por inoculación directa intracerebral, siendo esta última la más efectiva (se valora la infectividad como tiempo de incubación). La transmisión por sangre y derivados, aunque obtenida en condiciones de laboratorio, nunca ha podido ser demostrada en estudios epidemiológicos.
En ganado expuesto, en el caso del scrapie, la infectividad es detectada por primera vez en las placas de Peyer del íleon distal a los 6 meses de la exposición, sugiriendo una transmisión vía oral. La infectividad es detectada en el cerebro a los 2 años, y ésta va progresando lentamente con presentación de cambios espongiformes y manifestaciones clínicas durante el siguiente año6.
Para la mayoría de agentes causantes de EET, cuando son transmitidos por primera vez a otra especie, se alarga el tiempo de incubación (lo que se conoce como "barrera de especie"), tiempo que puede ser mayor que el tiempo de vida esperable para dicha especie. Con pases repetidos en la nueva especie, el tiempo de incubación decrece hasta acercarse al característico de la cepa causante y de la especie animal primaria7.
En la transmisión de las EET se ha demostrado la existencia de una susceptibilidad innata del individuo, más importante ésta que el grado de exposición. Este factor de susceptibilidad se asocia a la presencia de determinados genotipos del gen de la proteína priónica (PNRP).
Para explicar el proceso de muerte neuronal que acontece en las prionopatías se han establecido dos hipótesis, no necesariamente excluyentes. Se sabe que la isoforma proteasa-resistente, PrPsc, se acumula en tejido neural, tanto intracelularmente, en estructuras que son partes iniciales de vías endocíticas, como extracelularmente en general en forma de placas y amiloide. La primera hipótesis establece que esta acumulación interfiere con la función neuronal normal y causa su vacuolización y muerte7. La segunda hipótesis sugiere que es la ausencia del funcionalismo normal de la proteína priónica, PrPc, la que produce la muerte neu-
ronal.
Encefalopatías espongiformes transmisibles en animales
En los animales se conocen 6 EET: el scrapie, que afecta a ovejas y cabras; la encefalopatía transmisible de visones (ETV); la enfermedad caquectizante de alces y ciervos (ECC); la EET de rumiantes salvajes cautivos; la encefalopatía espongiforme felina, y la encefalopatía espongiforme bovina (EEB)1.
El scrapie, prototipo de enfermedad priónica, se trata de una enfermedad de distribución mundial, endémica en algunas regiones y es la primera prionopatía conocida, descrita en 1732. No existe evidencia de transmisión al ser humano del scrapie.
La epidemia de EEB, también denominada "enfermedad de las vacas locas", se detectó en el año 1986 en el Reino Unido8. El origen de la epidemia no está claro. Inicialmente se consideró que la enfermedad se había producido por la transmisión del scrapie ovino a la vaca a través de harinas animales; sin embargo, los estudios experimentales no han sido capaces de confirmar dicha hipótesis y no se descarta que el origen de la epidemia se encuentre en la entrada en la cadena alimentaria de casos de EEB esporádico, cuya existencia, por otro lado, tampoco ha podido ser demostrada. Sí está confirmado que el vector de transmisión fue la alimentación con harinas animales, coincidiendo, además la epidemia, con la modificación de la tecnología para obtener estos productos. La disminución de la temperatura y la supresión de los disolventes orgánicos para abaratar costes incrementó la probabilidad de persistencia de la infectividad9,10. Se cree que los primeros animales afectados fueron infectados durante el invierno de 1981 a 1982. Durante los siguientes años, el número de vacas afectadas fue aumentando de forma progresiva, de 16 casos en 1986 se pasó a 7.000 en 1989 y a 36.000 en 1992. Desde esta fecha, 5 años (es el tiempo considerado de incubación de la enfermedad) después de entrar en vigor en 1988 en el Reino Unido las normas que prohibían las prácticas previas de alimentación de rumiantes, el número de casos comenzó a disminuir, declinando desde entonces en una proporción del 40% al año. Posteriormente se han registrado casos de EEB en otros Estados europeos: Francia, Irlanda, Portugal, Suiza, Alemania, Países Bajos, España (en noviembre de 2000 se notifica el primer caso), Italia y Suecia.
En el Reino Unido se han descrito, asimismo, casos de encefalopatías espongiformes en gatos, así como de rumiantes exóticos que viven en cautividad, felinos salvajes y en mono rhesus alimentados con piensos animales11.
La infectividad en el ganado se ve limitada al tejido neural y linforreticular. No existe evidencia de infectividad en músculo o leche11. Tampoco se ha demostrado la existencia de una transmisión horizontal en ninguno de los más de 180.000 casos confirmados en más de 34.000 rebaños.
Encefalopatías espongiformes transmisibles en humanos
Las EET en el ser humano se pueden manifestar como un trastorno esporádico, hereditario o transmitido por medio de material exógeno que contenga priones. Han recibido el nombre de enfermedad de Creutzfeldt-Jakob (ECJ), nueva variante de ECJ (nvECJ), enfermedad de Gerstman-Straüssler-Scheinken (GSS), insomnio familiar letal (IFL) y kuru. La forma clinico-patológica más frecuente de EET es la ECJ, siendo la presentación esporádica o idiopática en un 85% de los casos, hereditaria en el 10-15%1-3, y iatrogénica en un porcentaje muy bajo1-3.
Enfermedad de Creutzfeldt-Jakob esporádica
La ECJ se describió por primera vez en los años veinte1. Se trata de una enfermedad de distribución universal, cuya incidencia anual se sitúa en 1-1,5 casos por millón de habitantes/año. No existe evidencia de cambios de incidencia en los últimos años y se ha descartado la existencia de una relación causal entre la epidemia de EEB y la ECJ esporádica12. Ninguno de los diferentes estudios de casos y controles ha identificado factores de riesgo para presentar la enfermedad13.
Características clínicas
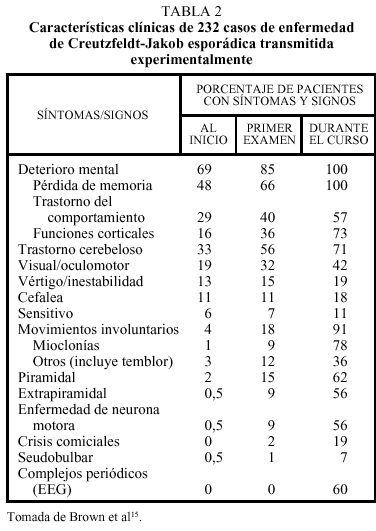
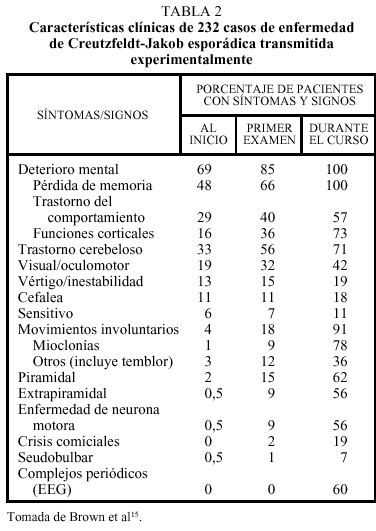
La enfermedad afecta por igual a varones y a mujeres. El intervalo de edad de inicio de la enfermedad se sitúa en 16-82 años, con un pico máximo a los 55-75, y una edad media de diagnóstico de 61,5 años. Aproximadamente un tercio de los pacientes presenta unos síntomas inespecíficos, que pueden considerarse prodrómicos, consistentes en astenia, anorexia, pérdida de peso y trastorno del sueño. En un tercio de los pacientes la enfermedad se manifiesta exclusivamente en forma de deterioro cognitivo, con pérdida de memoria, confusión o cambio del carácter; en otro tercio las manifestaciones fundamentalmente son en forma de ataxia cerebelosa o bien trastornos oculomotores o visuales; el tercio restante presenta una mezcla de estos síntomas (tabla 2)14,15.
En la mayoría de pacientes la forma de presentación de la enfermedad es subaguda, de semanas a meses, y sólo en un 20% de los casos el inicio es agudo. En los pacientes con el típico inicio subagudo, los fallos de memoria se hacen progresivos, con incapacidad para recordar eventos o nombres recientes, o presenta un síndrome confusional global. Los déficit en las funciones corticales se manifiestan en forma de incapacidad para encontrar la palabra adecuada, ejecutar cálculos aritméticos sencillos o escribir correctamente. Los síntomas más comunes de afección cerebelosa son ataxia de la marcha, vértigo y nistagmo, mientras que la ataxia de extremidades es menos frecuente. Los síntomas visuales consisten en diplopía, visión borrosa o distorsionada, alucinaciones o hemianopsia. La cefalea, las parestesias, los movimientos involuntarios y la presencia de signos de motoneurona inferior al inicio son infrecuentes. La clínica progresa y con frecuencia el fallecimiento se produce en un estado de mutismo acinético. La mayoría de los pacientes (90%) suele fallecer dentro del año del diagnóstico por una infección respiratoria o sistémica (la mitad de ellos en 5 meses), un 5% en el transcurso del segundo año y en el 5% restante la duración de la enfermedad es superior con casos que exceden los 10 años14,15. En estos pacientes con curso prolongado, el cuadro clínico de trastorno mental lentamente progresivo se sigue de un estadio terminal rápidamente progresivo de deterioro mental y físico típico de la forma subaguda de la enfermedad16.
Pruebas complementarias
Los hallazgos de laboratorio en sangre son normales. El líquido cefalorraquídeo (LCR) es acelular y con un contenido normal o ligeramente aumentado de proteínas (raramente superior a 100 mg/dl). En 1986, Hsich et al17 desarrollaron una técnica de inmunoblot para la detección de una proteína, la proteína 14-3-3 en LCR, que mostró ser altamente sensible y específica para el diagnóstico de la ECJ (fig. 2). La presencia de proteína 14-3-3 en LCR es un indicador inespecífico de destrucción grave neuronal aguda o subaguda, y los falsos positivos corresponden a pacientes con otros procesos de destrucción neuronal masiva (encefalitis herpética, ictus agudos, etc) o bien, dado que está también presente en hematíes, por contaminación hemática del LCR. Un estudio prospectivo multinacional de Zerr et al18 ha confirmado el alto valor diagnóstico de la prueba con una sensibilidad y especificidad del 91% para el diagnóstico de ECJ esporádica en un contexto clínico compatible. Sin embargo, esta prueba, por lo dicho, no es válida como test de cribado ante cualquier paciente con demencia o ataxia. En nuestro medio, Saiz et al19,20 también han demostrado la utilidad diagnóstica de la detección de la proteína 14-3-3 para el diagnóstico de la ECJ.
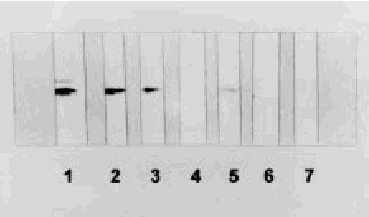
Fig. 2. Inmunoblot del líquido cefalorraquídeo (LCR) de los pacientes con enfermedad de Creutzfeldt-Jakob (ECJ) (tiras 2, 3 y 5) y de los pacientes con otras enfermedades neurológicas (tiras 4 y 6) que han sido inmunorreaccionados con un anticuerpo policlonal contra la proteína 14-3-3, demostrando una banda de movilidad electroforética idéntica a la encontrada en el control positivo que corresponde a un homogeneizado de cerebro de rata (tira 1). La tira 7 corresponde al LCR de un control negativo.
El electroencefalograma (EEG) es otro instrumento útil para el diagnóstico. En las fases iniciales de la enfermedad el EEG puede ser normal o mostrar un enlentecimiento inespecífico, pero su normalidad mantenida va en contra del diagnóstico de ECJ. En el curso de la enfermedad, las alteraciones se hacen más evidentes y se presentan los característicos complejos periódicos, bifásicos o trifásicos, sincronos y superpuestos al ritmo de base que está enlentecido21,22. Utilizando los criterios de Steinhoff21 su sensibilidad y especificidad son del 67% y 86%, respectivamente, para el diagnóstico de ECJ esporádica, y alcanza una sensibilidad del 90%22 con la realización de registros repetidos. Los complejos periódicos pueden detectarse, sin embargo, en otras encefalopatías, epilepsia, sobredosis por barbitúricos, intoxicación por litio, etc.
En la enfermedad avanzada la TAC craneal puede mostrar atrofia generalizada. En la RNM craneal, se puede observar hiperintensidad en T2 e intensidad protónica en ganglios basales en un 67% de los pacientes23.
Neuropatología
El examen macroscópico del encéfalo puede ser normal o mostrar atrofia global. Los hallazgos histológicos caracteríticos en la ECJ son la espongiosis, la pérdida neuronal, la astrocitosis y la formación de placas de amiloide (10% de casos esporádicos) (fig. 3)4. El cambio espongiforme consiste en una fina vacuolización del neurópilo de la sustancia gris. Se puede dar la confluencia de varias vacuolas constituyendo cavidades quísticas amplias. Mediante inmunohistoquímica, utilizando anticuerpos anti-PrPsc, se pueden poner de manifiesto los depósitos de PrPsc (fig. 4).
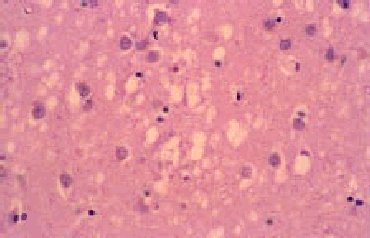
Fig. 3. Sección de encéfalo de un paciente con enfermedad de Creutzfeldt-Jakob, en la que se observa pérdida neuronal, microspongiosis confluente y astrocitosis. Tinción hematoxilina-eosina.
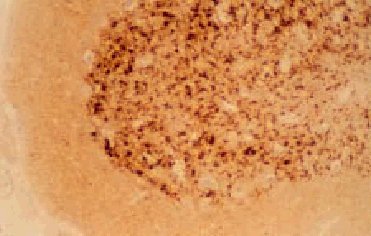
Fig. 4. Inmunohistoquímica de PrP. Se puede observar depósitos de PrP en una muestra de una paciente con enfermedad de Creutzfeldt-Jakob clásica esporádica.
Diagnóstico
El diagnóstico definitivo de la ECJ requiere el examen histológico de muestras de biopsia o autopsia que pon gan de manifiesto los hallazgos neuropatológicos característicos o la presencia de la PrPsc mediante inmunohistoquímica24. Para el diagnóstico en vida, la OMS ha establecido unos criterios en febrero de 199825, adaptados a partir de los originalmente propuestos por Masters et al26. Así los pacientes son diagnosticados de ECJ "probable" cuando presentan una demencia rápidamente progresiva de menos de 2 años de evolución, dos de los siguientes cuatro hallazgos: mioclonías, síntomas visuales y/o cerebelosos, signos piramidales y/o extrapiramidales, y mutismo acinético y presenten complejos peri ódicos en el EEG o positividad del test de proteína 14-3-3 en LCR. Aquellos pacientes que cumplen con todos los criterios, excepto por la presencia del EEG característico o positividad de 14-3-3, son clasificados como ECJ "posible" (tabla 3).

Polimorfismo del gen de la proteína priónica (PRNP)
El único factor identificado como indicador de susceptibilidad para ECJ esporádica es el genotipo de PRNP respecto al polimorfismo poblacional que este gen presenta en el codón 129. Así, un 85% de pacientes con ECJ esporádica son homozigotos para el codón 129, de los que de forma mayoritaria un 70-75% son metionina/metionina (M/M), y un 15% son heterozigotos (metionina/valina). Sin embargo, en la población general caucasiana, el genotipo M/M sólo está presente en el 37% de los sujetos. En las formas iatrogénicas de ECJ asociadas a la hormona de crecimiento, un 50% son V/V frente a sólo un 12% en la población general. Parece ser, por tanto, que el ser heterozigoto para el codón 129 confiere una protección parcial para presentar la enfermedad3.
El polimorfismo del codón 129 condiciona, además, el fenotipo clínico-patológico de ECJ, habiéndose descrito así en función del polimorfismo y del tipo de PrPsc 6 grupos de ECJ27. El fenotipo de ECJ clásico o variante mioclónica y la variante de Heindenhain se correlacionan con la presencia de homozigosis en el codón 129 y el patrón "tipo 1" de PrPsc.
Riesgos de infección
En la atención de estos pacientes se deben seguir las precauciones universales y no es necesario el aislamiento. No hay justificación para rechazar una autopsia o biopsia. En el manejo de tejidos deberán utilizarse guantes, delantales desechables, gafas y máscaras. Los instrumentos deberán de ser desechables o bien descontaminados mediante la exposición en hidróxido sódico 1 N (40 g/l) o hipoclorito sódico sin diluir durante una hora, y posteriormente en autoclave a 134 ºC durante una hora. Para la inactivación del prión en tejidos, se introducen éstos en ácido fórmico concentrado durante una hora, y posteriormente se mantienen en formol durante 48 horas28.
Nueva variedad de enfermedad de Creutzfeldt-Jakob
En abril de 199629 se publicaron los primeros 10 casos en el Reino Unido de una encefalopatía espongiforme humana con unas características clínicas y patológicas diferentes de las EET humanas previamentes conocidas y a la que se denominó nvECJ. Desde entonces y hasta enero de 2001 se han detectado 89 casos de nvECJ en el Reino Unido, uno en Irlanda y 3 en Francia.
La asociación temporal y geográfica con la EEB hicieron sospechar que la causa de la nueva variedad podía ser la infección por el agente de EEB. La exposición de la población humana al agente de la EEB probablemente fue máxima a finales de la década de los ochenta, antes de la aparición en 1988 de la prohibición del uso de despojos bovinos en la alimentación animal. Este dato es consistente con un período de incubación de 5-10 años. Esta sospecha epidemiológica fue posteriormente confirmada por la demostración de que macacos inoculados con EEB desarrollaban unas características patológicas similares a las de la nvECJ; por la presencia del tipo 4 de PrPsc en los casos de nvECJ, lo que la hace diferente de otras formas de ECJ, y que es similar a la encontrada en la EEB, y finalmente por la transmisión de la EEB a otras especies. Así, los agentes de la EEB, de la nvECJ y de la encefaloptía espongiforme de rumiantes exóticos y gatos muestran similares períodos de incubación y una distribución de las lesiones que difiere de la forma esporádica de ECJ. Aunque existen algunas variaciones entre casos, el fenotipo clínico de la nvECJ es relativamente uniforme y consistente con una única cepa de agente infeccioso. La vía de entrada es diges-
tiva, por consumo de productos cárnicos contamina dos con EEB. El prión penetraría en el organismo por las placas de Peyer intestinales y desde ahí se distri buiría por el sistema linforreticular, para posterior mente infectar el tejido neural. No se ha podido demostrar ninguna otra vía de infección diferente a la diges tiva.
Las características clínicas más prominentes de esta nueva variedad son: edad joven de presentación (mediana de 29 años), duración de la enfermedad de 9-35 meses y síntomas psiquiátricos, con frecuencia depresión, y síntomas sensitivos en forma de parestesias dolorosas como manifestaciones iniciales30. La ataxia cerebelosa se encontraba presente en todos los casos y al final de la enfermedad la mayoría presentaba mutismo acinético. Entre las pruebas complementarias, el EEG es anormal, pero no presenta la característica actividad periódica de la ECJ clásica; el test de proteína 14-3-3 es positivo en un 50% de los casos y la RM craneal presenta hiperintensidad en T2 en el tálamo posterior en un 70% de los casos32. El hallazgo neuropatológico más relevante, que define en la actualidad la nvECJ y la diferencia definitivamente de la forma clásica, es la presencia de placas de PrPsc con una morfología característica (placas floridas) ampliamente distribuidas por cerebro y cerebelo, y en menor número en ganglios basales, tálamo e hipotálamo. Ninguno de los pacientes con la nvECJ estudiados presenta mutaciones en el gen PRNP y todos ellos son homozigotos para metionina en el polimorfismo del codón 129.
El diagnóstico de la nvECJ se puede realizar en vida, incluso en el período presintomático, por estudio mediante inmunohistoquímica e inmunoblot de biopsia de amígdala palatina31 y no existen falsos positivos en otras enfermedades neurológicas. La afectación linforreticular (amígdala, nódulos linfáticos y bazo) encontrada en los casos de nvECJ, y no en otras formas de ECJ, parece ser clave en la patogenia priónica de esta nueva forma de ECJ.
Para el diagnóstico no histológico se han propuesto unos criterios32 basados en datos clínicos y pruebas complementarias con una sensibilidad del 64-77% y una especificidad del 100% para la categoría de probable (tabla 4).

Enfermedad de Creutzfeldt-Jakob iatrogénica
Hasta julio de 2000 se han notificado 267 casos de ECJ iatrogénica33 (tabla 5). Los casos iatrogénicos de ECJ han tenido lugar a través de diferentes fuentes y vías de adquisición: trasplante de córnea34, instrumentos neuroquirúrgicos26, electrodos implantados intracerebralmente, implantes de duramadre obtenidos de cadáveres1,16 y más de 100 casos distribuidos por todo el mundo relacionados con la administración de hormona del crecimiento y hormona gonadotrópica obtenida a partir de hipófisis de cadaveres humanos33. Esta última se presentó en Francia, donde el 4,4% de receptores manifestó la enfermedad, con una media de incubación de 12 años. No existe ninguna evidencia de que las transfusiones sanguíneas sean un factor de riesgo35; sin embargo, casos anecdóticos que habían recibido sangre han llevado a que se adopten precauciones al manejar productos sanguíneos de pacientes con ECJ, y que no se acepten donantes que hayan recibido hormona de crecimiento.

Encefalopatías espongiformes transmisibles genéticas
Un 10-15% de las personas con ECJ tiene una historia familiar compatible con una herencia autosómica dominante. En la mayoría de estos miembros se han detectado alguna de las 24 mutaciones causales descritas en la secuencia del gen de la proteína priónica (PRNP), situado en el brazo corto del cromosoma 203,6. En nuestro medio la mutación más prevalente es una mutación en el codón 200 (un cambio de glutamato por lisina: E200K), con un fenotipo prácticamente indiferenciable del de la ECJ esporádica.
El insomnio familiar letal (IFL) es un trastorno hereditario autosómico dominante asociado a una mutación del codón 178 con metionina en el codón 129 del alelo mutado36. El marcador patológico del IFL es la degeneración selectiva del talámo y de los núcleos olivares
inferiores. El primer paciente descrito presentaba un cuadro progresivo de insomnio intratable (de ahí su nombre), acompañado de disautonomía (hiperhidrosis, hipertermia, taquicardia e hipertensión), y en el curso de la enfermedad desarrolló alteraciones motoras, mioclonías, ataxia y demencia. Con posterioridad se ha descrito una gran variabilidad fenotípica en casos de IFL y la existencia de un solapamiento con ECJ3,37. Asimismo, se han comunicado casos de IFL sin la mutación característica, de origen esporádico.
En 1936, Gerstmann, Sträussler y Scheinker describieron un trastorno familiar, también de herencia autosómica dominante, caracterizado por ataxia cerebelosa progresiva acompañado de síntomas seudobulbares, disartria y paraparesia espástica38. La demencia se manifestaba tardíamente. El hallazgo neuropatológico más característico era la acumulación de placas de amiloide en cerebro y cerebelo. El curso suele ser prolongado, 5-11 años, aunque la edad media de fallecimiento es sólo de 48 años4. Un cambio de prolina por leucina en el codón 102 del PRNP es la mutación más frecuentemente asociada a GSS.
Debido a este solapamiento clínico existente entre distintas EET y la ausencia en algunos casos de historia familiar, la OMS recomienda la realización de estudio genético de PRNP a todo caso de EET, aun en ausencia de antecedentes familiares, o ante un cuadro neurodegenerativo no filiado por la posibilidad de que se trate de un caso genético de EET25.
Kuru
El kuru se describió en 195739. Su nombre significa "temblor" en la lengua original del grupo tribal Fore de Papúa-Nueva Guinea. El cuadro clínico se iniciaba con inestabilidad postural y temblor, que progresaban hasta que el paciente era incapaz de caminar sin ayuda. En las fases finales de la enfermedad se presentaba un trastorno cognitivo y el paciente moría en el plazo de un año. El hallazgo neuropatológico más característico era la presencia de placas de amiloide, "placas kuru", fundamentalmente en el cerebelo. En el pico de la epidemia la incidencia se situó en un 1% de la población. Se cree que la epidemia se inició y propagó por los ritos de canibalismo tras la muerte de un sujeto afectado de ECJ esporádica. Con la prohibición de este tipo de rituales en los años cincuenta, la enfermedad ha quedado virtualmente erradicada40.
Agradecimiento
Nuestro agradecimiento a la Dra. M. Jesús Rey (Banco de Tejidos Neurológicos) por proporcionar el material iconográfico sobre neuropatología y a Mercè Bonastre por su inestimable ayuda técnica.