






Los términos de hipoxia, anoxia e isquemia pueden llevar a alguna confusión. La hipoxia es el estado de reducción del oxígeno en un tejido con una adecuada perfusión. La anoxia es la ausencia de oxígeno con una adecuada perfusión también. La isquemia es la deprivación de oxígeno acompañada de una reducción de la perfusión en los tejidos.
Un infarto es una entidad morfológica que describe un área de tejido necrótico como resultado de una isquemia. De todos modos es evidente que el grado de isquemia tisular está determinado por un número de variables interacciones, como la adecuada circulación colateral suplente, las necesidades metabólicas del tejido perfusionado, o la existencia y la rapidez de una obstrucción. La interacción de estas variables es la razón que lleva a la isquemia miocárdica a presentarse de varias maneras: a) isquemia cardíaca crónica: angina estable, angina inestable (angina preinfarto) y angina variante (angina de Prinzmetal), y b) isquemia cardíaca aguda: infarto subendocárdico (sin ondas Q), transmural (con ondas Q) y muerte súbita.
En el campo del diagnóstico de esta enfermedad, sobre todo de la forma aguda, la medicina ha experimentado unas modificaciones recientes a las que el médico de asistencia primaria no puede ni debe ser ajeno, por lo que en esta revisión se va a hacer hincapié en los marcadores bioquímicos diagnósticos de esta patología.
Fisiopatología del infarto de miocardio
El infarto de miocardio suele ocurrir cuando disminuye bruscamente el flujo coronario después de una oclusión trombótica de una arteria ya estrechada por aterosclerosis. La estenosis de alto grado de las arterias coronarias de desarrollo lento no suelen precipitar un infarto agudo de miocardio, ya que con el tiempo se establece una generosa red colateral. En cambio, el infarto ocurre cuando se produce rápidamente un trombo en una arteria coronaria en una zona de lesión vascular (fisura). La lesión es producida o facilitada por factores tales como el tabaco, la hipertensión y el depósito de lípidos. Generalmente, el infarto sucede cuando la fisura rompe o ulcera la placa de ateroma y cuando las circunstancias (locales o sistémicas) favorecen la trombogénesis, de tal modo que se establece un trombo mural en el lugar de la rotura que ocluye la arteria coronaria. Una vez se establece la mucocapa inicial de plaquetas en la zona de rotura de la placa, diversos agonistas (colágeno, ADP, adrenalina, serotonina) inician la activación plaquetaria. Después de la estimulación por los agonistas, se observa una producción y liberación de tromboxano A2 (compuesto que induce vasoconstricción), continúa la activación plaquetaria y se establece una resistencia potencial a la trombólisis. La activación plaquetaria induce también un cambio en la conformación del receptor de la glucoproteína IIb-IIIa; este receptor tiene una gran afinidad con la cadena alfa del fibrinógeno al favorecer su entrecruzamiento y agregación. Asimismo, se activa la cascada de coagulación y los factores VII y X determinan, en última instancia, la conversión de la protrombina en trombina, la cual a su vez convierte el fibrinógeno en fibrina, y la arteria coronaria lesionada acaba obstruyéndose por un trombo que contiene agregados de plaquetas y bandas de fibrina (fig. 1).
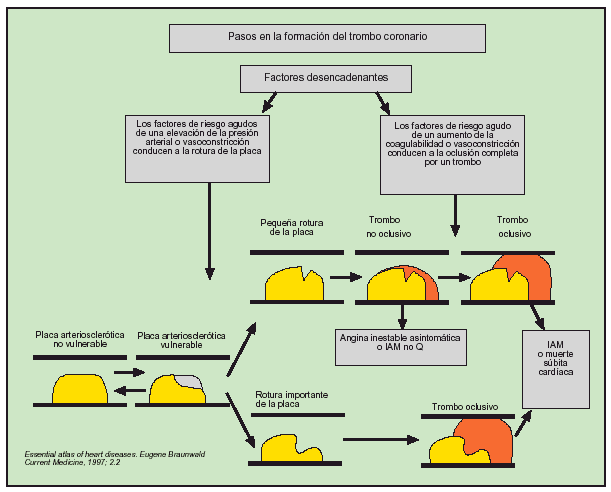
Fig. 1. Formación del trombo coronario.
Los enfermos con mayor riesgo de infarto de miocardio son los que tienen angina inestable o angina variante de Prinzmetal o bien varios factores de riesgo coronario. Algunas enfermedades menos comunes que predisponen al infarto son la hipercoagulabilidad, la colagenosis, el abuso de cocaína y los trombos o masas intracardíacas que provocan émbolos coronarios (fig. 2).
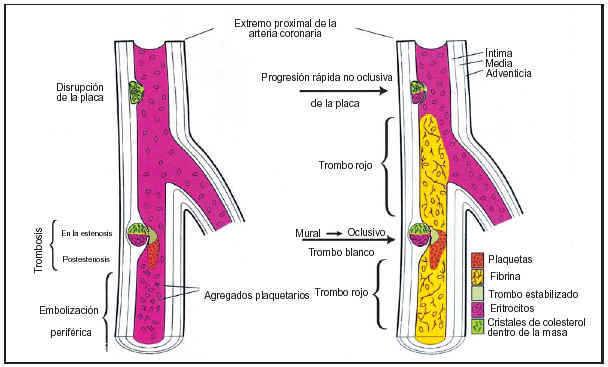
Fig. 2. Patogenia de la trombosis coronaria. La mayoría de los trombos coronarios (75%) son iniciados por la abertura plaquetaria. Exponiendo material trombogénico a la circulación sanguínea, siendo la pailla ateromatosa altamente trombogénica. El trombo tiene abundantes plaquetas y es de color gris claro en el punto de rotura. Si existe estenosis grave, se incrementa la trombosis por activación de las plaquetas inducida por el aumento del flujo. La fibrina, entonces, une las plaquetas estabilizando el trombo. La formación del trombo es dinámica: trombosis recurrente, trombólisis y embolización periférica ocurren simultáneamente con o sin vasospasmo concomitante, causando una obstrucción intermitente del flujo. La trombosis no oclusiva puede extenderse postestenóticamente, y si el trombo es rico en plaquetas puede ocluir el vaso, en este caso la sangre proximal y distal a la oclusión puede estancarse y coagularse, en sentido proximal o distal formando un trombo rojo de fibrina, parecido a una trombosis venosa. La propagación del trombo distal no ocluye las ramas principales.
Manifestaciones clínicas
En torno a la mitad de los pacientes que sufren infarto de miocardio se da algún factor desencadenante, como el estrés emocional grave, un ejercicio inusualmente intenso o una enfermedad grave. El dolor torácico es el síntoma prodrómico, que se prolonga más de 30 min, referido como opresivo, aplastante, constrictivo o como una tenaza, descrito por los enfermos como un puño cerrado contra el esternón, típicamente de localización retrosternal, que se extiende a la parte exterior del tórax, con frecuencia al lado izquierdo, y que a menudo irradia distalmente por los brazos, sobre todo el lado cubital del brazo izquierdo, a los hombros, las extremidades superiores, la mandíbula, el cuello y las regiones interescapulares y el epigastrio. Las náuseas, los vómitos y el dolor epigástrico son síntomas comunes que pueden llevar a confusión con trastornos gastrointestinales, sobre todo en ancianos y diabéticos. Este último grupo de pacientes puede no sufrir dolor torácico, y aparecen síntomas atípicos como mareo, palpitaciones, fatiga, síncope, aprensión, confusión o comienzo brusco de estado psicótico. Sorprendentemente, este tipo de pacientes puede tener un pronóstico grave igual que en la presentación convencional.
ECG
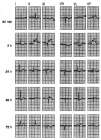
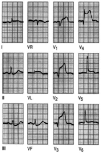
A menudo el primer signo electrocardiográfico de isquemia transmural aguda es el desarrollo de ondas T transitorias, gigantes, llamadas «hiperagudas», sobre el miocardio afectado. También se observa una elevación del segmento ST en las derivaciones que se encuentran sobre la isquemia cuando ésta afecta al epicardio, mientras que se produce depresión del segmento ST en presencia de isquemia subendocárdica. A los cambios hiperagudos de la onda T y las desviaciones del segmento ST le siguen inversiones de la onda T y a veces una pérdida de la altura de las ondas R y el desarrollo de ondas Q. Por el contrario, los infartos sin onda Q a menudo se acompañan de depresiones del segmento ST e inversiones de la onda T; este último sucede precozmente en el curso de un infarto y puede persistir después de que los segmentos ST deprimidos hayan regresado a la línea isoeléctrica en los pacientes que no reciben tratamiento de reperfusión; las elevaciones del segmento ST típicamente persisten durante 6-18 h, después del inicio del dolor torácico, y a medida que los segmentos ST se vuelven isoeléctricos las ondas T se invierten y se desarrollan ondas Q en las mismas derivaciones (figs. 3-8).
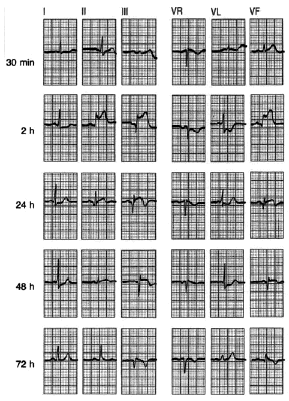
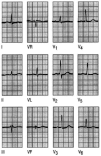
Fig. 3. Evolución del ECG de un infarto inferior. Inicialmente, el registro revela una onda T invertida en aVL, sin otra alteración significativa, más tarde, a las 2 h de inicio de los síntomas, se aprecia una elevación del segmento ST en las derivaciones correspondientes a esta localización, es decir, II, III y AVF. A las 24 h se evidencian las ondas T invertidas con gran claridad. A partir de este momento, y hasta las 48 h, se aprecian las ondas Q. A las 72 h, el segmento ST se vuelve a la línea isoeléctrica.
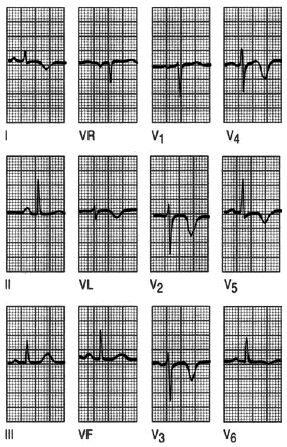
Fig. 4. Infarto sin onda Q. La ausencia de la onda Q, pero la presencia de ondas T invertidas en I, aVL y V2-V6 se deben a un infarto anterolateral sin ondas Q.
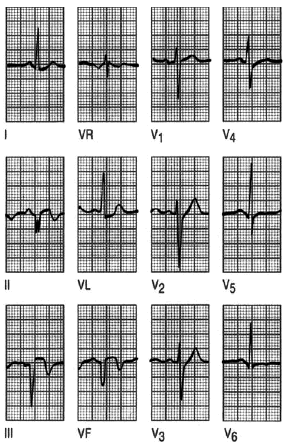
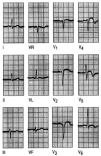
Fig. 5. Infarto de cara inferior. Cuando la pared inferior del ventrículo izquierdo está afectada, las alteraciones electrocardiográficas se observarán en las derivaciones inferiores, III, aVF y alguna vez en D II. Observa las ondas Q con segmento ST elevado y T invertida en las derivaciones II, III y aVF.
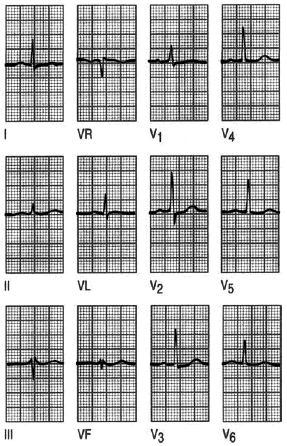
Fig. 6. Infarto posterior. El infarto posterior, algunos autores lo denominan «verdadero posterior», porque en la antigua terminología, lo que ahora se denomina inferior, se refería al infarto posterior. La onda R dominante del «verdadero» posterior, causa un ECG de apariencia similar a la hipertrofia ventricular derecha, pero no se acompaña en este caso con desviación del eje. Aquí es la sintomatología del paciente, la falta de hipertrofia en el examen físico y, sobre todo, los marcadores serológicos lo que determina el diagnóstico.
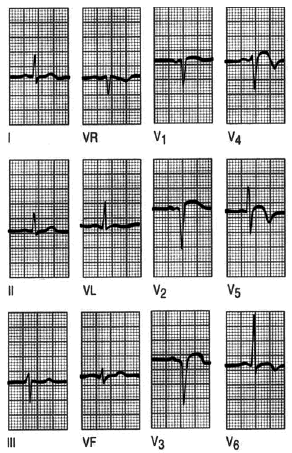
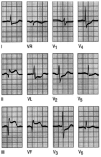
Fig. 7. Infarto de cara anterior. El registro es normal en las derivaciones de los miembros, sólo llama la atención una onda T plana en aVL. Hay ondas Q en V1-V3 y sólo una R inadecuada en V4. El ST está elevado en V2-V5. La T está invertida en V4-V6. La elevación del segmento ST sugiere un infarto agudo, pero la presencia de ondas Q indica que ha ocurrido al menos hace una hora o dos.
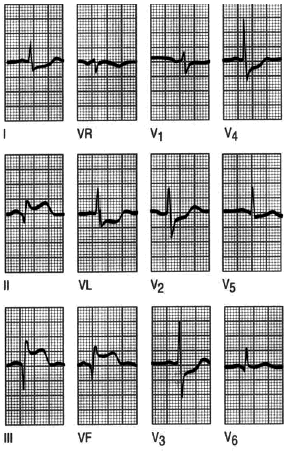
Fig. 8. Infarto de cara inferior. Un registro de un paciente con los primeros síntomas de dolor precordial, que demuestra un infarto agudo de miocardio de cara inferior con ondas Q y segmento ST elevado en las derivaciones correspondientes a esta localización. Se observa la depresión del segmento ST en las derivaciones precordiales V2-V5. Este hallazgo se denomina alguna vez depresión recíproca, y se presenta en la isquemia en el territorio afectado por el infarto. La evidencia de la enfermedad en dos territorios, en este caso el inferior y el anterior, es signo de mal pronóstico.
Durante muchos años los infartos con ondas Q se llamaron infartos transmurales y los infartos sin ondas Q infartos no transmurales. Sin embargo, la correlación anatomopatológica-ECG no apoya esta terminología y las denominaciones que se prefieren en la actualidad son simplemente infartos con onda Q e infartos sin onda Q.
El infarto puede estar localizado en la región septal anterior del ventrículo izquierdo si los cambios del ECG mencionados anteriormente se producen en las derivaciones V1 a V3, en el ápex del ventrículo izquierdo si se producen en las derivaciones V4 a V6, en la pared lateral con cambios en las derivaciones V5, V6 y aVL, y en la pared inferior con cambios en las derivaciones II, III y aVF. El infarto de la pared posterior puede causar una depresión recíproca del segmento ST y una elevación paradójica de la onda R en las derivaciones V1 a V4. El infarto del ventrículo derecho produce una desviación del segmento ST y un patrón QS en las derivaciones de la derecha (V1, V3R y V4R) y, generalmente, se acompañan de infarto de la pared inferior.
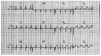
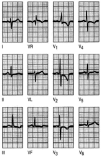
En una minoría de pacientes con infarto de miocardio el ECG es completamente normal o presenta sólo pequeños cambios en el segmento ST y la onda T, de ahí la importancia de los marcadores bioquímicos en la cardiopatía isquémica aguda (figs. 9 y 10).
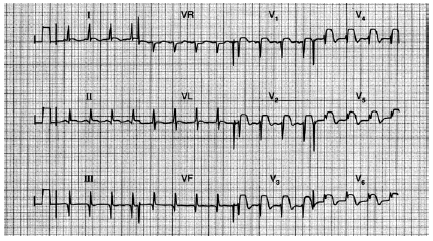
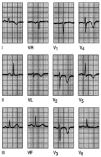
Fig. 9. Registro de paciente de 60 años de edad que hace 3 años sufrió un IAM de cara inferior tras una angina después de exposición al frío; fue derivado al hospital con dolor precordial después de una hora al no responder a nitratos sublinguales. Obsérvese las ondas Q en las derivaciones II, III y aVF, así como los complejos QRS en las derivaciones anteriores, con una elevación del segmento ST en V1 a V6.Este registro evidencia un infarto antiguo de cara inferior con infar to agudo de miocardio de cara anterior.
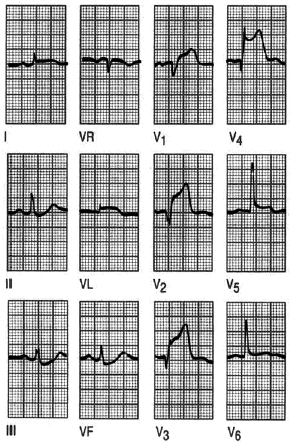
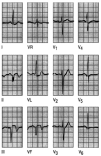
Fig. 10. Infarto anterolateral. Elevación del segmento ST en I, aVL, V1 a V6. Obsérvense las ondas Q en V2 a V4.
Marcadores bioquímicos
Lactato deshidrogenasa (LDH)
En los últimos años ha perdido protagonismo como marcador serológico del infarto de miocardio, ya que ha sido desplazado por otros más fiables y operativos. Pero como todavía persiste en muchos libros de texto actuales y dado el papel fundamental que desempeñó en épocas pasadas, hemos creído conveniente recordarlo. Realmente, el gran interés actual en isoenzimología deriva de las observaciones de las múltiples formas moleculares de la LDH. Las isoenzimas de la LDH se designan en la práctica de acuerdo con su movilidad electroforética. La fracción con mayor movilidad se denomina LDH1, la que tiene menos movilidad se conoce como LDH5 y las otras tres se designan como LDH2, LDH3 y LDH4, respectivamente. La LDH del miocardio, de los hematíes y del riñón contienen gran parte de isoenzima de movilidad rápida (LDH1 y LDH2). Después del infarto de miocardio, la actividad sérica de la LDH aumenta entre 2 y 4 veces y persiste dicha elevación durante un tiempo considerable (entre 10 y 14 días); la determinación de las isoenzimas de LDH proporciona una mayor fiabilidad. Sin embargo, la «LDH invertida» (LDH1 > LDH2) es un fenómeno más variable que puede resultar evidente a las 12 h y detectarse dentro de las primeras 48 h aproximadamente en el 20% de los pacientes con infarto de miocardio.
La utilización simultánea de las determinaciones de las isoenzimas de la CPK y la LDH combina el alto grado de sensibilidad derivado de la CPK con el alto grado de especificidad de las isoenzimas de LDH.
Los criterios combinados se cumplen en los especímenes obtenidos durante las primeras 48 h después de un episodio agudo de posible ataque al miocardio. Se observa en los pacientes con CK2 (MB) un patrón propio de LDH invertida (LDH1 > LDH2).
Mioglobina
Aumenta rápidamente 1-2 h después de la lesión muscular, y es secretada fuera de la sangre antes de 12-24 h por el riñón. Por tanto, es un buen marcador temprano en caso de una buena función renal y en ausencia de traumatismo del musculosqueleto, inyecciones intramusculares, ejercicio físico intenso y ausencia de toxinas o fármacos.
El valor plasmático normal de la mioglobina en el suero es inferior a 90 μg/l. Es una molécula relativamente pequeña que se excreta de forma rápida por la orina y, por tanto, la duración de su evolución tras el infarto es menor de 24 h. Aunque la mioglobina es un marcador muy sensible de necrosis tisular, es inespecífica porque es un constituyente del musculosqueleto y puede liberarse de este tejido incluso con una ligera lesión. No tiene estructura cardioespecífica. Así, muchos autores no la recomiendan como un marcador diagnóstico. Sin embargo, otros estudios han confirmado que la mioglobina es mejor marcador que la CPK-MB y la troponina T para excluir el infarto de miocardio en las primeras 6 h de su presentación debido a su rápida liberación, casi en la primera hora de la lesión muscular; así pues, en ausencia de lesión del musculosqueleto o de fallo renal, el test de la mioglobina es una gran ayuda en el diagnóstico o la exclusión del infarto agudo de miocardio en su fase más temprana. En un estudio prospectivo de la mioglobina en la evolución temprana del dolor agudo precordial, ésta tenía una sensibilidad del 100% en el diagnóstico de infarto de miocardio en las primeras 2 h, mejor que la CPK-MB debida a los falsos positivos de esta última5. La mioglobina ha sido también propuesta como un índice útil para medir la perfusión satisfactoria y poder evaluar el tamaño del infarto.
Creatinfosfocinasa (CPK)
Esta enzima se concentra de forma elevada en el musculosqueleto y en el miocardio. Se encuentra también en el cerebro en cantidades apreciables, mientras que en otros órganos sólo hay pequeñas cantidades y está ausente en el hígado. Se eleva en las siguientes situaciones: infarto de miocardio, daño en el musculosqueleto, ejercicio físico intenso, inyecciones intramusculares, reacciones psicóticas agudas, infarto pulmonar y edema pulmonar (en estudios recientes).
Isoenzimas de la creatincinasa
Las propiedades fisioquímicas de la creatincinasa detectadas en los extractos de corazón, cerebro y musculosqueleto humano son diferentes: la enzima cerebral (CK1) y la enzima del musculosqueleto (CK3).
La enzima CPK-MB es la isoenzima que se encuentra en gran concentración en el músculo cardíaco, pero no es exclusiva ya que una proporción se encuentra en el musculosqueleto. Esta proporción no está bien definida en la bibliografía médica actual, mientras que unos autores indican que el 60% pertenece al miocardio y el 40% al musculosqueleto y otros señalan un 70% frente al 30%, de todos modos y como «mínimo común múltiplo» es aceptado por todos la gran proporción del miocardio frente al musculosqueleto. Así la relación (índice relativo) entre la CPK-MB y la CPK total > 2,5 sugiere una elevación miocárdica más que esquelética de la CPK-MB pero no es diagnóstica. Incluso esta relación queda totalmente en entredicho y es inútil cuando los valores de la CPK total aumentan a causa de una lesión del musculosqueleto o cuando los valores de la CPK total son normales pero se eleva la CPK-MB. La inquietud del clínico respecto al hallazgo de un marcador más fiable que la CPK-MB llevó a perseguir la forma miocárdica de la CPK-MB, y se constató que esta enzima (CPK-MB2) es atacada en la circulación por la enzima carboxipeptidasa, que escinde un residuo lisina del extremo carboxídico para dar una isoforma (CPK-MB1) con movilidad electroforética distinta. Una relación CPK-MB2/CPK-MB1 > 1,5 indica un infarto de miocardio con gran sensibilidad, sobre todo si han transcurrido 4-6 h desde la obstrucción coronaria.
El complejo de las troponinas
Este complejo contiene tres moléculas: troponina C (p.m.: 18KDa), troponina I (p.m.: 26KDa) y troponina T (p.m.: 39KDa) (fig. 11).
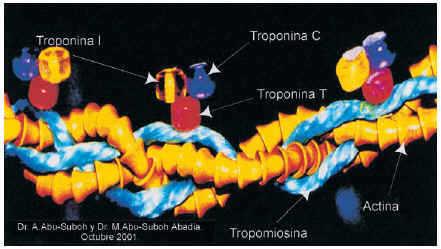
Fig. 11. Presentación en maqueta del complejo de las troponinas, donde se aprecia la tropomiosina, una proteína alargada parecida a una varilla que se enrosca alrededor del filamento de la actina, dotándolo de estabilidad y rigidez. La tropomiosina no tiene utilidad clínica. El complejo de las troponinas es el encargado de la fijación de la actina a la miosina. Esta estructura se encuentra fijada a la molécula de la tropomiosina y está formada por tres polipéptidos distintos denominados T, I y C. La troponina T une el complejo a la molécula de la tropomiosina situado en el complejo en el punto del filamento de la actina donde se producirá la interacción con la miosina. La troponina I impide físicamente la unión de la miosina a la actina. La troponina C fija iones de calcio que inducen el cambio conformacional en el complejo de troponinas permitiendo que la miosina pueda estar en contacto con el filamento de la actina. La troponina detecta el daño miocárdico con más sensibilidad que la CPK-MB y permite el diagnóstico de «microinfartos» que ocurren a veces en la angina inestable. Además, la troponina T y la troponina I cardioespecífica se mantienen elevadas, sobre todo la primera, durante más de una semana, lo que permite el diagnóstico de infarto, incluso si éste ha sucedido con un intervalo mayor a 48 h desde el episodio hasta que el paciente acude a la consulta. La troponina I y la troponina T constituyen la prueba de elección y han sustituido a la LDH y sus isoenzimas utilizadas en otra época.
Este complejo está situado en los filamentos finos del músculo estriado contráctil. Cada una de estas moléculas está codificada por un gen diferente. La troponina C interviene en la contracción muscular dependiendo de la concentración del calcio. Esta troponina no tiene una estructura cardíaca específica; así, el ascenso de la concentración sanguínea de troponina C puede estar tanto en la lesión muscular esquelética como en la cardíaca, por lo que no tiene interés como marcador en la cardiopatía isquémica.
La troponina I cardioespecífica demuestra un 40% de diferencia con la troponina I musculosquelética y, además, contiene 31 aminoácidos residuales en su N-terminal no presentes en la forma esquelética. Estas diferencias han permitido el desarrollo de anticuerpos monoclonales para los ensayos de troponina I cardioespecífica. La troponina T presenta diferencias en la forma musculosquelética y cardíaca en unos 6-11 aminoácidos residuales, que permitió su distinción por anticuerpos monoclonales.
La concentración de la troponina T e I cardioespecíficas en el suero es de 0-0,1 y 1-3 μg/l, respectivamente. La concentración de ambas empieza a incrementarse a las 4-8 h después de la lesión miocárdica y llega a su pico máximo a las 12-24 h. La troponina T puede permanecer elevada más allá de las 2 semanas y la troponina I permanece elevada más de 5-7 días, lo cual ha permitido escogerlas como marcadores excelentes en el diagnóstico serológico del infarto de miocardio. Tenemos que tener en cuenta que éstas pueden elevarse en enfermedades musculares, pero la rápida determinación de estas troponinas a la cabecera del paciente les confiere un lugar prioritario en el diagnóstico serológico de la cardiopatía isquémica; incluso se han detectado microinfartos en las anginas inestables donde no puede elevarse la CPK-MB.
La troponina T se eleva mucho más que la troponina I en pacientes con enfermedad renal crónica y hemodializados, y en ausencia de enfermedad isquémica miocárdica, diabetes mellitus o enfermedad muscular. McLauren et al lo demostraron en un grupo de 24 pacientes en diálisis y sin cardiopatía isquémica. De este grupo, un 71% presentaba una elevación de la troponina T, frente al 4% que presentaba una elevación de la troponina I. Otros trabajos han ido reforzando este hecho, pues es evidente que en este grupo de pacientes la troponina I es más fiable como marcador en caso de cardiopatía isquémica aguda; no obstante, este tema es de debate actual. Por último, la relación no es específica entre el daño miocárdico del infarto y la leucocitosis polimorfonuclear que aparece pocas horas después de los primeros síntomas y persiste entre 3 y 7 días, llegando a las cifras de 12.000-15.000 leucocitos/μl. La VSG aumenta más lentamente de lo que cabría esperar para la cifra de leucocitos, se eleva a lo largo de la primera semana y persiste durante una o dos semanas.
Diagnóstico diferencial del infarto agudo de miocardio
Una historia clínica, un ECG típico y la liberación en el suero de un marcador macromolecular como la CPK-Mb o la troponina T son los tres pilares para establecer el diagnóstico de IAM. La OMS requiere la presencia de dos de estos tres hallazgos para su diagnóstico. Aunque los pacientes con angina inestable pueden tener síntomas y cambios del segmento ST y la onda T en el ECG similares a los del IAM, se pueden distinguir a posteriori por la ausencia de desarrollo de nuevas ondas Q y la falta de liberación de marcadores macromoleculares en el suero. La diferenciación del IAM de la pericarditis aguda puede ser difícil porque el dolor y las elevaciones del segmento ST pueden ser similares y los marcadores séricos a veces se elevan ligeramente en la pericarditis, el dolor a menudo persiste durante varios días, las elevaciones del segmento ST son típicamente más persistentes y extensas que en el IAM (generalmente, más de 6 de las 12 derivaciones), acompañadas muchas veces con depresión del segmento PR; además, los segmentos ST permanecen elevados durante días después de que las ondas T se hayan invertido y no se desarrollan ondas Q. La característica del dolor difiere por ligero alivio al inclinar al paciente hacia delante en la pericarditis (figs. 12-14).
Fig. 12. Embolia pulmonar múltiple. Nótese la desviación del eje a la derecha, el dominio de la onda R en V1, la presencia de onda S en V6. T invertida en V1-V4.
Fig. 13. En este registro hay una elevación del ST en DI, aVL y de V2 a V5. El paciente presentaba un cuadro clínico de infarto de miocardio, y éste debe ser también el diagnóstico. No hay, sin embargo, ondas Q, por lo que a partir del ECG, exclusivamente, no se puede hacer un diagnóstico de certeza. En este caso, la determinación de los marcadores bioquímicos serán de gran ayuda.
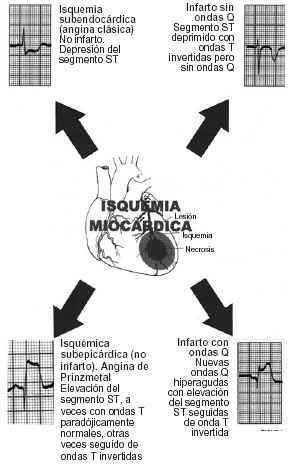
Fig. 14. Diferentes aspectos electrocardiográficos de la cardiopatía isquémica.
Otros diagnósticos y procedimientos que pueden cursar con elevación de enzimas miocárdicas
Cardioversión
La mayoría de los estudios están de acuerdo en que la cardioversión eleva la actividad de la CPK total y la CPK-MB. O'Neill et al demostraron que el 50% de estos pacientes sufre una alteración de esas enzimas hasta 100 veces el valor de referencia después de administrar 6 o más aplicaciones. Aunque la mayoría pertenece al musculosqueleto, en pocos pacientes también aumentarán las enzimas cardíacas. La conclusión de estos trabajos es que la alteración enzimática después de una sesión de cardioversión puede minimizar los cambios encontrados en el infarto de miocardio, aunque éstos son de naturaleza transitoria y usualmente vuelven a la línea de base antes de los 15 min desde la última administración.
Inyecciones intramusculares
La CPK total puede aumentar hasta 15 veces su actividad en las 6 h después de una inyección intramuscular. La CPK-MB no experimenta aumento. Este hecho tiene que tenerse en cuenta en pacientes con dolor precordial, para no administrar analgesia por vía intramuscular a fin de no alterar los marcadores enzimáticos.
Tromboembolismo pulmonar
El embolismo pulmonar es la tercera causa más común de las enfermedades cardiovasculares. A pesar de que la analítica enzimática determina la isquemia aguda miocárdica, ésta aporta muy poca ayuda en el embolismo pulmonar excepto para excluir otras entidades. Esta enfermedad se denomina The great masquerade («La gran enmascarada»), porque el diagnóstico diferencial incluye el infarto de miocardio, la neumonía, la insuficiencia cardíaca congestiva, la fractura de costilla, el neumotórax y otras enfermedades graves en las que están presentes dolor torácico, pleurítico y fatiga. Cuando el infarto de miocardio, la neumonía o el fallo cardíaco congestivo no responden al tratamiento, la coexistencia de un tromboembolismo pulmonar debe ser tenido en cuenta. El embolismo pulmonar tiene como patrón la relativa tardanza de la elevación de la CPK total en comparación con la precocidad de los signos de la mioglobina y la CPK-MB.
Abuso de cocaína
Hay que tener presentes las complicaciones cardíacas del abuso de la cocaína u otras drogas sintéticas nuevas. La cocaína, en concreto, bloquea los receptores de la adrenalina y la noradrenalina, aumenta la reserva de noradrenalina y puede estimular el aumento de las catecolaminas desde la glándula adrenal. Como resultado de ello, la cocaína causa taquicardia, hipertensión y vasoconstricción (espasmo de las arterias coronarias epicárdicas) llevando a la isquemia, el infarto o la muerte súbita. Estos efectos pueden ser potencialmente exagerados en pacientes con cardiopatía coronaria previa, aunque sea estable; asimismo, este efecto vasoconstrictor puede ser exagerado con el uso concomitante del tabaco o del alcohol. Como la cocaína puede inducir isquemia miocárdica, infarto o disección aórtica, el dolor torácico de los consumidores de esta droga debe ser estudiado en profundidad, investigando los metabolitos urinarios de la cocaína, a modo de test, ante la sospecha de su consumo en pacientes con esta sintomatología precordiálgica. La totalidad de la CPK y la CPK-MB están elevadas por la rabdiomiólisis, causada por esta sustancia, aunque a veces encontramos que la CPK-MB permanece invariable. De hecho, muchos autores constatan esta controversia de la CPK-MB, es decir, entre los pacientes consumidores de cocaína y con dolor torácico miocárdico, unos presentan una elevación de esta enzima mientras otros no lo hacen. En estos casos el diagnóstico electrocardiográfico y las troponinas son suficientes para clasificar el cuadro de infarto agudo de miocardio.
Miocarditis
La miocarditis puede ser aguda o crónica. Es un proceso inflamatorio que afecta al corazón. Puede ser debido a una amplia gama de infecciones (sobre todo virales), cuadros tóxicos o daño inmunológicos o hipersensibilidad medicamentosa. Como cualquier proceso inflamatorio, la miocarditis puede presentar una gama amplia, desde un estado asintomático, un estado de infección aguda hasta un estado fulminante, incluso con fatal desenlace. Una pequeña proporción de los pacientes puede presentar una figura mímica de un infarto de miocardio. La CPK total puede incrementarse hasta 10 veces el límite de referencia con la CPK-MB, excediendo en ocasiones los valores límite hasta el 40%. Estos casos presentan un desafío diagnóstico considerable, que muchas veces requiere investigaciones serológicas minuciosas inmunológicas, y a menudo precisan biopsia. Investigaciones recientes han sugerido que la troponina T cardioespecífica es una ayuda estimable para la valoración de la implicación cardíaca.
Pericarditis
Es un síndrome inflamatorio caracterizado por un dolor pericárdico y características específicas en el ECG (elevación del segmento ST en más de 6 de las 12 derivaciones y depresión del segmento PR en dichas derivaciones, que objetivan una elevación del segmento ST). El síndrome puede ser causado por una amplia gama de condiciones, como en la miocarditis, y puede ocurrir después del infarto de miocardio. Es aceptado, generalmente, que la mayoría de los pacientes con una infección aguda que presentan los cambios típicos de la pericarditis aguda pueden tener alteraciones de la CPK y la CPK-MB. La característica principal de este patrón es la rápida normalización de estas enzimas, coincidiendo con la normalización del ECG. Estos hallazgos han sugerido una lesión de corta duración de las células miocárdicas.
Polimiositis
Es una inflamación difusa que afecta al musculosqueleto proximal. Cuando la enfermedad afecta también la piel se denomina dermatomiositis. Hay un interés creciente de la afección cardíaca de esta enfermedad. Por causa de la afección muscular esquelética hay un incremento de la CPK total, pero la involucración del músculo cardíaco ha sugerido que la CPK-MB es superior al 5% de la CPK total. El creciente uso de la troponina T ha demostrado un daño inequívoco del músculo cardíaco en esta enfermedad. El aumento de la troponina T hasta 100 veces el límite de referencia se encontró en el 27% de los pacientes con polimiositis/dermomiositis.
Estrategia a seguir en la investigación del infarto agudo de miocardio
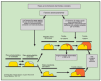
A. Ralph Henderson, del Royal Hospital de Londres, sugiere el siguiente protocolo en la investigación de pacientes con sospecha de infarto de miocardio, que ha sido seguido por varios hospitales de tercer nivel en Estados Unidos y Europa, incluido nuestro país. Así, se ha agrupado a los pacientes en cuatro categorías con respecto a los primeros síntomas a su llegada al hospital: a) los que llegan antes de 6 h; b) los que llegan en el período de 6-12 h; c) los que llegan en el período de 12-24 h, y d) los que llegan pasadas las 24 h.
Pacientes que llegan antes de 6 h
Pacientes que llegan antes de 3 h
ECG diagnóstico de infarto de miocardio. El diagnóstico es clínico, no requiere pruebas analíticas. Si la clínica no es convincente, se debe realizar el test de mioglobina al llegar. Si la mioglobina es positiva, hay que realizar el test de CPK-MB en el período de 3-6 h. Si la mioglobina es negativa, hay que volver a realizar el mismo test 2-4 h más tarde y confirmarlo con la CPK-MB.
ECG dudoso. Realizar la mioglobina al llegar. Si la mioglobina es positiva realizar CPK-MB en el período de 3-6 h. Si la mioglobina es negativa volver a hacer el test y confirmarlo con la CPK-MB.
Pacientes que llegan entre 3 y 6 h antes
ECG diagnóstico de infarto de miocardio. El diagnóstico es clínico y no precisa pruebas. Si hay dudas hay que practicar analítica: mioglobina y CPK-MB al llegar:
Confirmar los resultados positivos con troponina T (o I), determinación que debe realizarse en el período de 6-9 h.
Si la mioglobina y la CPK son negativas, hay que volver a hacer las pruebas 2-4 h más tarde, confirmándolo con la troponina T o I.
ECG no diagnóstico de infarto. Practicar mioglobina y CPK-MB2 al llegar, confirmar los resultados positivos con troponina T o I en 6-9 h, si la mioglobina y la CPK-MB son negativas volver a repetir 2-4 h más tarde, y confirmarlo con la troponina T o la troponina I.
Pacientes que llegan entre 6 y 12 h antes
ECG diagnóstico de infarto
No precisa más pruebas. Si existen dudas: practicar CPK-MB al llegar y confirmar los resultados positivos con la troponina T o la I.
ECG no diagnóstico
Practicar CPK-MB al llegar, y si es negativo repetir en las próximas 2-4 h.
Confirmar los resultados positivos con troponina T o con troponina I.
Pacientes que llegan en el período de 12-24 h
ECG diagnóstico
Diagnóstico clínico, no precisa más pruebas. Si existen dudas practicar CPK-MB al llegar, y si es negativo repetir pasadas 2-4 h.
Confirmar la CPK-MB positiva con la determinación de la troponina T o la troponina I.
ECG no diagnóstico
Determinar las CPK-MB al llegar, y repetir a las 2-4 h si es negativa, confirmar las CPK-MB positivas con la troponina T o la troponina I.
Pacientes que llegan más tarde de las 24 h
Tanto si el ECG es diagnóstico como si no, hay que determinar la troponina T o la troponina I al llegar y 4-8 h más tarde repetir la determinación para confirmar el hallazgo inicial.
Tratamiento prehospitalario
El mayor riesgo para la vida se produce durante las primeras horas después de la oclusión coronaria y disminuye progresivamente en el tiempo posterior. En efecto, aproximadamente la mitad de las muertes asociadas con el infarto agudo de miocardio se producen durante las primeras horas después del inicio y son debidas, generalmente, a una fibrilación ventricular. Por tanto, el principio fundamental en la actuación ante el infarto es acortar el tiempo entre el inicio de los síntomas y el tratamiento a un mínimo absoluto. Los tres componentes de la demora en el inicio del tratamiento son: a) demora del paciente en buscar atención médica; b) evaluación prehospitalaria y traslado, y c) evaluación e inicio del tratamiento en el hospital. Se debe dirigir la atención a la reducción de cada uno de estos componentes.
Se debe informar a la población general, especialmente a los pacientes con riesgo elevado de infarto o reinfarto, sobre los síntomas prodrómicos del infarto. Es útil para los que tienen un riesgo elevado, como los que han sufrido previamente un síndrome coronario agudo (IAM o angina inestable), llevar siempre con ellos una copia de su ECG en reposo para servir de base a futuras comparaciones. Es deseable que los servicios médicos de urgencia capacitados apliquen la desfibrilación ventricular, ya que si los pacientes con IAM son desfibrilados inmediatamente, su supervivencia y recuperación son excelentes. Los cuidados prehospitalarios deben incluir el establecimiento de un acceso venoso, calmar enseguida el dolor con 5 mg de sulfato de morfina cada 5 min 3 veces si es necesario, la administración de nitroglicerina y el tratamiento de la taquicardia ventricular con lidocaína (un bolo de 75-100 mg seguido de una perfusión de 1-2 mg/min).
Trombólisis prehospitalaria
Dado que es de gran importancia el restablecimiento de la reperfusión coronaria lo más precozmente posible, el tratamiento trombolítico debe comenzar cuanto antes en los pacientes que lo necesitan. Cuando el tratamiento prehospitalario permite iniciar el tratamiento trombolítico más de una hora antes que en el hospital, dicho tratamiento iniciado prehospitalariamente puede reducir la mortalidad relativa en un 15-20% por debajo de la que se alcanza con el tratamiento estándar iniciado en el hospital. El tratamiento trombolítico prehospitalario puede ser especialmente útil en comunidades en las que la demora por transporte es de 90 min o más. Para la realización de la trombólisis prehospitalaria, no obstante, se requieren médicos, o más comúnmente técnicos en urgencias médicas, muy bien formados y con experiencia en las ambulancias, así como medios técnicos para transmitir el ECG electrónicamente al hospital receptor. Si no, es mejor potenciar el transporte rápido y el acortamiento del tiempo de instauración del tratamiento ya en urgencias.
Elección del agente trombolítico
Ha habido un considerable debate acerca de la elección del agente trombolítico. Actualmente, han sido aprobados la estreptocinasa, la t-PA (activasa), la r-PA (reteplasa) y el complejo activador del plasminógeno de la estriptocinasa anisoilado (APSAC).
A los pacientes tratados con t-PA o r-PA se les debe administrar heparina intravenosa (1 bolo de 60-70 U/ kg/h) durante aproximadamente 48 h. El tiempo de tromboplastina parcial activado (TTPa) se debe medir inicialmente 6 h después del inicio de la trombólisis, y después al menos cada 24 h, haciendo ajustes para mantener un TTPa de 50-75 s. A los pacientes que reciben estreptocinasa se les debe agregar heparina si hay un gran infarto de pared anterior, insuficiencia cardíaca o un trombo mural visible en la ecocardiografía.
Observaciones sobre el tratamiento médico del infarto de miocardio
El mayor beneficio en cuanto a mortalidad del tratamiento crónico con bloqueadores beta se observa en pacientes con deterioro de la función auricular y arritmias ventriculares. Los bloqueadores beta se deben administrar indefinidamente en pacientes con IAM confirmado. La aspirina, a dosis de 160 o 325 mg/día, se debe administrar indefinidamente. El tratamiento con un inhibidor de la ECA durante un período indefinido se recomienda en pacientes con indicios de insuficiencia cardíaca congestiva, una fracción de eyección por debajo del 40% o una gran anomalía regional del movimiento de la pared.
Se debe considerar la restitución de estrógenos en las mujeres posmenopáusicas, con postinfarto sin riesgos mayores que los usuales de cáncer de mama (una historia familiar de cáncer de mama). Este tratamiento mejora el perfil lipídico; hay estudios de observación epidemiológica que han demostrado una reducción en los episodios coronarios con este tratamiento. Las mujeres posmenopáusicas a las que se les ha practicado una histerectomía, generalmente, se tratan con 0,625 mg de estrógenos conjugados orales/día. En las que tienen útero, se emplea una combinación de estrógenos y progesterona.
Los fármacos antiarrítmicos no deben utilizarse como profilaxis de rutina en el paciente postinfarto. Los que tienen taquiarritmias ventriculares graves sintomáticas se deben tratar con bloqueadores beta, se deben someter a detección de isquemia provocable y considerar la posibilidad de aplicaciones especializadas.
Bases del tratamiento farmacológico del infarto de miocardio
Bloqueadores betaadrenérgicos
Estos fármacos disminuyen el consumo miocárdico de oxígeno, reduciendo la frecuencia cardíaca, la presión arterial y la contractibilidad miocárdica. Diversos ensayos multicéntricos han demostrado los beneficios del tratamiento precoz, es decir, menos de 4 h después del inicio del dolor, con bloqueadores beta intravenosos seguidos de tratamiento oral. Los pacientes que acuden más tarde pueden iniciar el tratamiento oral, que generalmente se continúa durante un período indefinido.
Cuando se administra de forma i.v. en la etapa aguda, el tratamiento bloqueador beta ha demostrado reducir el desarrollo del IAM definitivo, disminuir el tamaño del infarto y la incidencia de taquiarritmias, muerte súbita y mortalidad total. Cuando se administran crónicamente, estos fármacos reducen la incidencia de reinfarto, muerte súbita y mortalidad total. Si se desarrollan complicaciones como insuficiencia cardíaca, bloqueo cardíaco o broncospasmo, los bloqueadores beta se deben retirar o disminuir las dosis hasta que se resuelven las complicaciones.
Contraindicaciones al bloqueo betaadrenérgico agudo. Frecuencia cardíaca menor de 60 lat/min, presión arterial sistólica menor de 100 mmHg, insuficiencia ventricular izquierda moderada o grave, signos de hipoperfusión periférica, intervalo PR mayor de 0,24 s, bloqueo AV de segundo o tercer grado, enfermedad pulmonar obstructiva grave, historia de asma, enfermedad vascular periférica grave, diabetes mellitus tipo 1.
Dos regímenes útiles de esta familia son: atenolol i.v. de 5 a 10 seguido de atenolol oral, 100 mg/día, o metoprolol 15 mg i.v. divididos en tres dosis, seguidos de 100 mg orales dos veces por día.
Inhibidores de la ECA
Por medio de la disminución de la poscarga y la precarga ventricular, los inhibidores de la ECA reducen la incidencia de la insuficiencia cardíaca congestiva, la dilatación ventricular y la remodelación. Estos fármacos parecen ser útiles cuando se administran durante un mes, comenzando pronto, un día después del infarto. Cuando se continúan durante varios años, mejoran el resultado clínico en pacientes con disfunción ventricular e insuficiencia cardíaca. En ausencia de complicaciones (hipotensión, hipersensibilidad conocida, estenosis bilateral de las arterias renales y posible embarazo), se debe administrar un inhibidor de la ECA oral a todos los pacientes con IAM hemodinámicamente estables con elevación del segmento ST.
El tratamiento con IECA se debe empezar en las primera 24 h, después de que se haya completado un tratamiento de perfusión y la presión arterial se haya estabilizado.
Se debe evaluar la función ventricular izquierda antes del alta hospitalaria, y la inhibición de la ECA se debe continuar durante un período indefinido en pacientes con insuficiencia cardíaca, con infarto previo, con indicios de fracción de eyección reducida (< 40%) o con anomalías extensas del movimiento regional de la pared. En pacientes sin ninguno de estos rasgos en el momento del alta hospitalaria, se puede interrumpir el IECA. Se pueden emplear varios regímenes, como lisinopril 5 mg/día durante 2 días, y después 10 mg/día, o trandolapril 2 mg/día durante 2 días, seguidos de 4 mg/día.
Nitroglicerina
Este fármaco ejerce acciones farmacológicas favorables porque reduce la precarga y la poscarga de los ventrículos derecho e izquierdo y produce vasodilatación coronaria. No hay indicios claros de que la administración sistemática de la nitroglicerina sea beneficiosa en pacientes con IAM no complicado que reciben tratamiento trombólico. Sin embargo, la nitroglicerina i.v. (de 5-10 µg/min a 150 μg/min hasta la estabilidad hemodinámica) es el fármaco de elección en la recurrencia de la angina. Es especialmente útil en pacientes no hipotensos con insuficiencia ventricular izquierda, y se puede utilizar durante 24-48 h en pacientes con IAM extenso de la pared anterior con onda Q para reducir la remodelación y la dilatación ventriculares. Después se puede continuar de forma oral o tópica durante 4-6 días en estos pacientes.
Antagonistas del calcio
No se ha demostrado que estos fármacos reduzcan la mortalidad en el IAM. De hecho, dado que se ha indicado que el nifedipino de liberación inmediata se asocia con un riesgo de mortalidad intrahospitalaria que aumenta en relación con la dosis, este fármaco no se debe utilizar en pacientes con IAM. El verapamilo (240-480 mg/día en dosis divididas, o una vez al día con liberación sostenida) o el diltiazem (120-360 mg/día en tres o cuatro dosis o una vez al día con liberación sostenida) pueden ser útiles para controlar los ataques de isquemia recurrente o fibrilación auricular en pacientes sin insuficiencia cardíaca ni bloqueo AV.
Cuidados después del alta
El médico de atención primaria desempeña un papel fundamental en los cuidados después del alta y en la prevención del reinfarto. Quizá lo más importante sea la educación respecto a la naturaleza de la enfermedad arterial coronaria y los factores de riesgo del infarto recurrente. Las modificaciones del estilo de vida tienen una importancia fundamental. Entre ellos cabe destacar el abandono del tabaco; se debe remarcar al paciente que 2 años después de dejar de fumar, el riesgo de un infarto recurrente no mortal disminuye al límite observado en los que nunca fumaron. El médico de atención primaria debe programar al paciente postinfarto para la rehabilitación cardíaca, se deben llevar a cabo programas de adiestramiento físico; caminar en terreno llano, ir en bicicleta y realizar técnicas de calentamiento suave que se deben incrementar progresivamente. Una prueba de esfuerzo limitada por síntomas negativos a las 3-6 semanas después del alta se debe tomar como señal para aumentar la actividad física e incluir natación, marcha rápida y jogging. Es útil que el ejercicio se lleve a cabo como parte de un programa organizado de rehabilitación, que incluya instrucciones dietéticas, conseguir un peso óptimo y el abandono del tabaco. A los pacientes con una prueba de esfuerzo limitada por síntomas ligeramente anormales a las 3-6 semanas, se les debe animar a hacer ejercicio físico pero no a un ritmo que provoque regularmente síntomas de isquemia.
Bibliografía general
Alpert JS. Cardiology for the primary care. Philadelphia: Mosby, 2000.
Bayés de Luna A. Electrocardiografía clínica. Barcelona: Harcourt Brace de España, S.A., 1996.
Braunwald E. Essential atlas of heart diseases. Philadelphia: Appleton & Lange, 1997.
Braunwald E, Hauser SL, Fauci AS, Longo DL, Kasper DL, Jameson JL. Principles of internal medicine. 15th ed. San Francisco: McGraw-Hill, 2001.
Brown DL. Cardiac intensive care. San Diego: WB Saunders Company, 1998.
Califf RM. Atlas of heart diseases. Acute miocardial infarction and other acute ischemic syndromes. Volume VIII. Philadelphia: Current Medicine, 1996.
Castellano Reyes C. Urgencias cardiovasculares, Tomo 1. Madrid: Harcourt Brace, 1999.
Chatterjee K. Cardiology and illustrated text/reference. Volume 2. Philadelphia: J.B. Lippincott Company, 1991.
Fleming JS. Atlas electrocardiografía práctica. 2.a ed. Barcelona: Ediciones Doyma, 1986.
Goldman L, Braunwald E. Cardiología en atención primaria. Madrid: Harcout, 2000.
Hampton JR. The ECG in practice. 3th ed. New York: Churchill Livingstone, 1997.
Henry JB. Diagnóstico y tratamientos clínicos por el laboratorio. 9.a ed. Masson, S.A., 2000.
Maroto Montero JM, De Pablo Zarzosa C, Artigao Ramírez R, Morales Durán D. Rehabilitación cardíaca. Barcelona: Olalla Cardiología, 1999.
Maynard SJ, Menown IB, Adgey AAJ. Troponin T or troponin I as cardiac markers in ischaemic heart disease. Heart 2000;83:371-3.
Moss DW. Enzyme tests in diagnoses. London: Arnold, 1998.
PcPhee SJ. Pathophysiology of disease. An introduction to clinical medicine. 3th ed. San Francisco: Lange Medical Books/McGraw-Hill, 2000.
Price SA, Wilson LM. Pathophysiology, clinical concepts of disease processes. 5th ed. St. Louis, Missouri: Mosby, 1997.
Rude P, et al. Electrocardiographic and clinical criteria for recognition of acute miocardial infartation based on analysis of 3,697 patients. Am J Cardiol 1983;52:936-42.
Stevens A, Lowe J. Texto y atlas de histología. Barcelona: Doyma, 1993.
Wagner GS. Marriot's practical electrocardiography. 10th ed. Philadelphia: Lippincott Williams & Wilkins, 2001.