






Las enfermedades cardiovasculares son la principal causa de muerte en los países desarrollados. Representan el 20% de la mortalidad global en el mundo1. La intervención sobre los factores de riesgo tradicionales y el tratamiento clásico de la cardiopatía isquémica no han conseguido revertir las dramáticas consecuencias de esta enfermedad. En los últimos años la investigación se ha centrado en la etiopatogenia de la aterosclerosis con el objeto de encontrar nuevas dianas terapéuticas que nos ayuden a mejorar el control de la enfermedad aterosclerótica.
Desde que en 1988 Saikku et al2 publicaron su ya clásico trabajo seroepidemiológico en el que encontraron una asociación entre cardiopatía isquémica y serología positiva frente a Chlamydia pneumoniae, se ha intentado demostrar la relación etiopatogénica entre la infección y la cardiopatía isquémica. Aunque pueda parecer novedosa esta tendencia, no lo es. Ya en el año 1908, sir William Osler sugirió una posible asociación entre la infección y la aterosclerosis. Asimismo, en 1911, Frothingham3 afirmaba que la esclerosis de la edad podría ser una suma de factores que irían desde procesos infecciosos hasta tóxicos o metabólicos.
Se sabe desde hace tiempo que los factores de riesgo clásicos de cardiopatía isquémica (sexo, edad, tabaco, hipercolesterolemia, hipertensión arterial, diabetes mellitus) sólo explican el 50% de los casos de aterosclerosis clínicamente aparentes1. De igual forma, pacientes con niveles similares de riesgo cardiovascular experimentan cursos diferentes de la enfermedad y también es conocido el hecho de que las tasas de infarto de miocardio y muerte súbita se incrementan en invierno y tras epidemias de gripe4. Todos estos hallazgos han soportado la hipótesis de que las infecciones podrían condicionar un mayor riesgo de aterosclerosis.
En los últimos años se han descubierto determinados agentes infecciosos relacionados con diversas enfermedades; de ellas la más aceptada quizá sea la enfermedad ulcerosa con el Helicobacter pylori. De hecho, en el tratamiento de la úlcera péptica es indicación erradicar el Helicobacter mediante un tratamiento con antibióticos asociado a un inhibidor de la bomba de protones. Otras enfermedades relacionadas con agentes infecciosos pueden verse en la tabla 1.

La primera evidencia consistente que asoció aterosclerosis e infección fue publicada por Paterson y Cottral5 en 1940. Estos autores observaron una fuerte asociación entre el virus de la enfermedad de Mareck (virus de la familia herpesviridae) y la aterosclerosis. Tras infectar pollos con este virus, demostraron que los animales infectados presentaban lesiones anatomopatológicas en la pared arterial similares a las descritas en la arteriosclerosis humana.
Aterosclerosis. Concepto
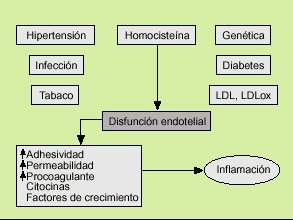
Las primeras teorías que intentan explicar la aterosclerosis datan de 1852, cuando Von Rokitansky formuló la hipótesis de la incrustación. Esta hipótesis defendía que el fenómeno central del proceso aterosclerótico era el depósito de fibrina en las células de la pared arterial, y este hecho condicionaría la posterior acumulación de lípidos en la arteria. Pocos años después, en 1856, Virchow6 formuló la hipótesis lipídica que se basaba en que el flujo sanguíneo, mediante las fuerzas de cizallamiento, entre otros factores, ocasionaba un daño en el endotelio que determinaba el acúmulo de lípidos en la pared arterial. Esta teoría ha permanecido vigente durante más de un siglo, de forma que el evento nuclear en la aterosclerosis era el depósito lipídico, fundamentalmente de colesterol. En la década de los ochenta se desarrolla el modelo actual promulgado por Ross7, en el que se considera a la aterosclerosis como un fenómeno inflamatorio. La hipótesis de respuesta a la lesión, defendida por Ross, especula con que diversos agentes nocivos, como agresiones mecánicas, químicas, víricas o metabólicas, provocan una disfunción endotelial. La célula endotelial desempeña un papel fundamental en la homeostasis del flujo sanguíneo. Su alteración va a provocar una secreción de diversas sustancias, como citocinas, factores de crecimiento, etc., que van a inclinar la balanza hacia el estado protrombótico y van a atraer a los monocitos/macrófagos y a los linfocitos que van a desencadenar y perpetuar el fenómeno inflamatorio8.
Los factores de riesgo cardiovascular clásicos (hipertensión arterial, diabetes mellitus, hipercolesterolemia, tabaco, homocisteína, predisposición genética) y otros más recientes, como determinadas infecciones, provocan una disfunción endotelial que desencadenará el fenómeno aterosclerótico. El endotelio, en respuesta a estas agresiones, va a sufrir una serie de cambios. Se favorece la expresión de moléculas de adhesión en su superficie (ICAM-1, VCAM-1), que junto con un aumento de la permeabilidad traduce un paso de monocitos/macrófagos y leucocitos al intersticio vascular. Paralelamente la producción de óxido nítrico y de prostaciclina por el endotelio se atenúa, lo que determina una vasoconstricción y una mayor adhesividad plaquetaria. A su vez la célula endotelial es capaz de secretar citocinas y factores de crecimiento que junto a los fenómenos anteriores provoca una inflamación en el intersticio de la pared arterial (fig. 1).
La respuesta inflamatoria está mediada por las sustancias producidas por los macrófagos y linfocitos T. Estas sustancias (factor de crecimiento derivado de las plaquetas, factor de crecimiento de los fibroblastos, etc.) provocan una migración de células musculares lisas y su posterior proliferación. Unas células musculares lisas conservan su función contráctil y otras la pierden y son capaces de producir matriz extracelular, constituyendo la capa fibrosa que envuelve al núcleo lipídico. Los monocitos al atravesar la pared arterial se diferencian en macrófagos y sintetizan una gran cantidad de sustancias que se van a encargar de perpetuar la respuesta inflamatoria y estimular el sistema inmune. Una de sus funciones principales es fagocitar a las lipoproteínas de baja densidad (LDL), acción mediada por las LDL oxidadas (LDL-ox), la interleucina 1 y el factor de necrosis tumoral alfa (TNF-*), entre otras, constituyendo las células espumosas que son el sustrato anatómico del núcleo lipídico de la placa de ateroma. Los linfocitos T son atraídos por la acción del TNF-*, la interleucina 2 y el factor estimulante de colonias de los granulocitos/macrófagos. Éstos son los grandes desconocidos de todo el proceso, y actualmente una de las múltiples líneas de investigación se centra en intentar esclarecer su papel en el proceso aterosclerótico. Se sabe que los linfocitos T cooperadores (CD4) predominan en las placas maduras y que los linfocitos T supresores (CD8) predominan en las lesiones precursoras de la placa o en la periferia de las lesiones9. Su papel no está todavía bien definido.
El proceso que precede a la formación de la placa de ateroma es la disfunción endotelial. El primer hallazgo anatomopatológico descrito son las estrías grasas, que consisten en la acumulación de macrófagos repletos de vesículas lipídicas en su interior, conteniendo el exceso de LDL fagocitadas (células espumosas), junto con linfocitos T activados. Esta lesión es común en niños y adolescentes y traduce el inicio de la inflamación en la pared arterial. Posteriormente se produce la migración de las células musculares lisas que envuelven a esta lesión aislándola de la luz arterial y constituyen la capa fibrosa. En el interior se van a acumular los linfocitos T estimulados y las células espumosas junto a detritus celulares y necrosis celular. Esta lesión constituye la placa estable, que no suele producir clínica hasta tiempo después, cuando su ruptura la transforma en placa inestable. La ruptura de la placa de ateroma es el desencadenante de los fenómenos coronarios agudos. Hasta que la placa no ocupa el 50% del diámetro arterial no incide de forma importante en la luz vascular10. Los fenómenos coronarios agudos no se producen por la oclusión completa de la luz por la placa de ateroma, puesto que este fenómeno suele ser tardío. Antes se produce la ruptura de la placa, exponiendo la matriz extracelular al flujo sanguíneo, lo que condiciona la adhesión y formación de un trombo que, al ocluir la luz, va a provocar la isquemia distal a la lesión. La ruptura de la placa suele producirse por la acción de metaloproteasas, como las colagenasas producidas por los macrófagos activados de la placa que van adelgazando la capa fibrosa8.
Aterosclerosis e infección
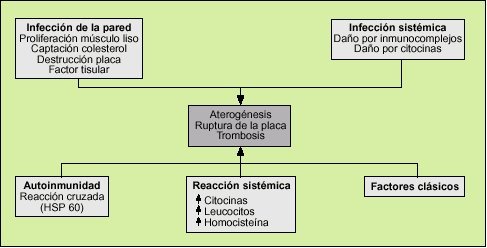
Las infecciones actuarían como un factor de riesgo más en el desarrollo de la aterosclerosis. Son muchas las hipótesis que intentan explicar esta asociación (fig. 2).
Efectos directos sobre la pared arterial
Proliferación de células musculares lisas
Cuando se infecta una célula por citomegalovirus (CMV) se producen cambios en la misma que conducen a su muerte. Cuando estos cambios no se producen se habla de infección abortiva. Experimentalmente esta forma de infección puede conseguirse infectando una célula con una cepa de CMV que tiene tropismo para infectar células de otra especie, por ejemplo, infectar células humanas con CMV obtenido de células de pollo infectadas. La infección abortiva de CMV induce la expresión de los productos de genes inmediatos, como IE2-84, que se unen al p53 inhibiendo su acción y provoca que la célula no finalice el ciclo celular11. La infección abortiva por CMV inhibe el fenómeno de apoptosis necesario para el recambio celular en los tejidos.
Acúmulo de lípidos en la pared arterial
Se ha demostrado que las infecciones por herpes virus disminuyen la actividad lisosómica y citoplásmica de la hidrólisis de colesterol, provocando su acumulación en la placa de ateroma12. Asimismo otro de los productos de los genes inmediatos del CMV provocados por una infección abortiva (IE1-72) aumenta la actividad del receptor encargado de depurar las lipoproteínas de baja densidad oxidadas13. La infección del endotelio por el CMV induce la expresión de las moléculas de adhesión en la superficie de la célula endotelial.
Efectos directos sistémicos de la infección
Los fenómenos de respuesta inflamatoria frente a cualquier agente infeccioso pueden provocar daño endotelial que precipite la aterogénesis. La formación de inmunocomplejos y la síntesis de citocinas pueden dañar el endotelio directamente.
Efectos indirectos de la infección
Los procesos infecciosos generan una respuesta sistémica cuya expresión es la elevación de los reactantes de fase aguda (proteína C reactiva, etc.), la leucocitosis y la secreción de múltiples mediadores químicos, como las citocinas producidas por los macrófagos, que condicionan una respuesta inflamatoria general que puede alterar diversos sistemas y, entre ellos, favorecer un estado procoagulante. Los factores secretados por los macrófagos activados son, fundamentalmente, la interleucina 1, la interleucina 6, el interferón * y el TNF-*. El TNF-* disminuye la actividad de la lipoproteinlipasa, por lo que aumentan los niveles de triglicéridos y disminuyen las lipoproteínas de alta densidad (HDL)14. Algunos microorganismos pueden estimular directamente la producción de citocinas. El lipopolisacárido de C. pneumoniae estimula la producción de las interleucinas 1 y 6, del TNF-* y favorece la expresión en la superficie endotelial de las moléculas de adhesión.
Fenómenos de autoinmunidad
Se postula que parte del daño endotelial está mediado por una reacción cruzada entre anticuerpos contra antígenos bacterianos con una gran homología con antígenos humanos. En determinadas situaciones de estrés se sintetizan ciertas proteínas denominadas HSP (heat shock proteins) para proteger a las proteínas celulares de ser desnaturalizadas. Se ha demostrado que estas proteínas se han conservado a lo largo de la cadena evolutiva y, por ejemplo, la HSP 60 de C. pneumoniae y de H. pylori15 es muy similar a la expresada en el endotelio humano en situaciones de estrés. La respuesta inmunológica frente a estos determinantes antigénicos podría causar daño endotelial16.
Además de todas estas consideraciones, hay que tener en cuenta que la variabilidad de cada individuo puede condicionar una diferente respuesta a estas agresiones. Según la respuesta inflamatoria que desencadene el individuo, junto a la respuesta inmune frente a los diferentes patógenos, existirá una mayor o menor susceptibilidad a la formación de aterosclerosis por un estímulo infeccioso.
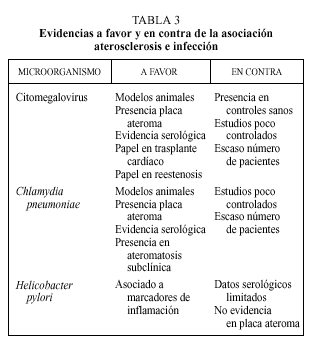
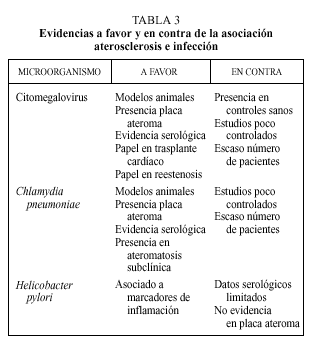
En la génesis de la aterosclerosis se han involucrado a diferentes microorganismos (tabla 2). De entre todos ellos, el CMV, C. pneumoniae y H. pylori son sobre los que existe una evidencia mayor. A continuación se desarrollan los argumentos a favor y en contra de la asociación de estos microorganismos con la aterosclerosis. La evidencia científica de esta asociación se sustenta en cuatro tipos de estudios:

1) Estudios seroepidemiológicos.
2) Estudios patológicos. Demostración del patógeno dentro de la placa de ateroma.
3) Existencia de modelos animales y experimentos in vitro.
4) Estudios de intervención terapéutica.
Aterosclerosis y Helicobacter pylori
El H. pylori es una bacteria microaerofílica, gramnegativa, de forma espiral, que reside en las células de la mucosa gástrica y que se transmite por un mecanismo fecal-oral. Está presente en prácticamente el 100% de las úlceras duodenales y en el 60% de las úlceras gástricas. La infección por H. pylori se adquiere habitualmente en la infancia y los anticuerpos frente a ella persistirán hasta edades avanzadas de la vida, por lo que
la prevalencia de anticuerpos frente a Helicobacter en la población general es muy alta, alrededor del 40% a los 50 años de edad17. La prevalencia de estos anticuerpos se correlaciona con la edad y el estatus socioeconómico. Hasta la fecha no hay ningún ensayo de intervención terapéutica ni existen modelos animales que expliquen la asociación con la aterosclerosis, por lo que ésta se va a fundamentar en estudios seroepidemiológicos y en algunos estudios patológicos.
Estudios seroepidemiológicos
Numerosos estudios han confirmado una asociación entre serología positiva frente a H. pylori y enfermedad aterosclerótica18-20. La mayoría de ellos se han realizado en pacientes con cardiopatía isquémica clásica y algunos sobre aterosclerosis cerebral. En un metaanálisis de trece artículos21 se encontró una odds ratio (OR) de 2,11 (intervalo de confianza [CI] entre 1,21 y 3,69). No todos los estudios han demostrado una asociación positiva y los que apoyan esta asociación tienen en unos casos una OR en torno a 1 y en otros los intervalos de confianza son excesivamente amplios debido al escaso número de pacientes incluidos en la mayoría de los estudios. No hemos encontrado ningún estudio con más de 100 pacientes que muestre una asociación positiva.
En general no se puede concluir que las evidencias encontradas sean convincentes, puesto que la mayoría presentan diversas limitaciones. Hay pocos estudios prospectivos y la mayoría son estudios caso/control que plantean una mayor dificultad metodológica en el control de posibles sesgos. Casi todos están realizados con un número limitado de casos y controles, por lo que los resultados son difícilmente extrapolables a la población general y los intervalos de confianza son excesivamente amplios, lo que resta fiabilidad a los resultados. Su principal inconveniente es el escaso ajuste para posibles factores de confusión. La mayoría ajustan en función de la edad y el sexo; algunos, además, tienen en cuenta en su análisis algunos de los factores de riesgo clásicos de cardiopatía isquémica, pero pocos ajustan frente al estatus socioeconómico. Se sabe que tanto la infección por H. pylori como la cardiopatía isquémica son más frecuentes en pacientes con inferior nivel social y económico. La falta de homogeneidad en las poblaciones de los distintos estudios hace extraordinariamente difícil establecer comparaciones entre ellos.
Estudios patológicos
Hasta el momento ningún estudio con suficiente número de pacientes ha demostrado la presencia de la bacteria en la pared arterial22. Este hecho podría ser explicable por los estrictos requerimientos de la bacteria para su crecimiento, ya que requiere un medio con un pH bastante ácido para sobrevivir.
En algún estudio se ha correlacionado la serología positiva para H. pylori con una mayor severidad de las lesiones observadas en las arterias coronarias mediante coronariografías23.
A pesar de los inconvenientes para aislar la bacteria en el torrente sanguíneo y en la pared arterial se han postulado varias teorías que intentan explicar esta asociación. Algunos estudios aseguran que la infección por H. pylori se asocia a un aumento de reactantes de fase aguda como la proteína C reactiva y el fibrinógeno24. Asimismo se ha observado que el tratamiento erradicador del Helicobacter se correlacionaba con una disminución de los niveles de fibrinógeno mayor25. Estudios aislados parecen relacionar la infección con una alteración del metabolismo lipídico que produce una disminución de las HDL y una elevación de los triglicéridos26.
Otro de los mecanismos de daño endotelial podría ser una reactividad cruzada frente a las HSP 60 del Helicobacter, que tienen una gran homología con las HSP del endotelio humano15. Por último, un soporte a esta relación puede venir del aumento de homocisteína secundario a la infección. La presencia de Helicobacter en la mucosa gástrica provocaría una gastritis crónica que va a condicionar una disminución en la absorción de ácido fólico, vitamina B6 y vitamina B1227. Estas vitaminas intervienen en el metabolismo de la metionina y su disminución condiciona que no se catalicen las reacciones enzimáticas para el paso de homocisteína a metionina, lo que provocaría un aumento de la homocisteína que influiría en el daño endotelial.
Aterosclerosis y citomegalovirus
Los primeros agentes infecciosos relacionados con la aterosclerosis fueron los virus, y de todos ellos, la familia de los herpesvirus. El origen de estas teorías proviene de los experimentos de Fabricant28, quien demostró que al inyectar un virus de la familia Herpesviridae (virus de la enfermedad de Mareck) en pollos alimentados con una dieta rica en colesterol se desarrollaban lesiones ateroscleróticas a lo largo de toda la pared arterial, más intensas que en aquellos no infectados. Posteriormente, el grupo de Hajjar12 demostró un importante acúmulo de ésteres de colesterol en las células musculares lisas humanas infectadas por el virus del herpes simple. Sin embargo, ulteriores estudios seroepidemiológicos no han podido demostrar una asociación significativa entre serología positiva frente a virus herpes simple y la aterosclerosis17, quedando únicamente la asociación entre el CMV y la enfermedad aterosclerótica.
El CMV es un virus de distribución universal que pertenece a la familia Herpesviridae. Se puede transmitir a través de vía fecal-oral, oral-oral o parenteral. Se cree que su hábitat natural son los leucocitos, aunque esto no está bien establecido. La infección en individuos inmunocompetentes permanece en estado latente a lo largo de la vida, aunque puede sufrir reactivaciones, asintomáticas en la mayoría de los casos. La prevalencia en la población general es muy elevada; más del 50% de la población mayor de 35 años tiene anticuerpos frente al virus, y conforme aumenta la edad estas cifras se elevan hasta alcanzar un 70% en los mayores de 70 años. La infección por CMV se correlaciona con un estatus socioeconómico bajo, con estados de inmunosupresión y con la edad.
Estudios seroepidemiológicos
La mayoría de los estudios demuestran una asociación positiva entre infección por CMV y la aterosclerosis17 con una OR > 2. Es importante destacar que hay pocos estudios en cardiopatía isquémica clásica con el CMV, puesto que la mayoría están hechos en receptores de trasplante cardíaco, en estudios de reestenosis tras angioplastia y en la aterosclerosis carotídea17. Los pacientes con serología positiva frente a CMV parece que con el tiempo tienen un mayor riesgo de desarrollar aterosclerosis carotídea. Sin embargo, dada la alta prevalencia de serología positiva en la población general y la dificultad para ajustar en función de los múltiples factores de confusión, no se puede establecer esta asociación con certeza.
Una de las asociaciones más citadas es la de la infección por CMV en la reestenosis de las arterias coronarias tras angioplastia. Se estima una tasa de reestenosis de un 30%, aunque esta complicación se ha reducido con la colocación de stents tras la angioplastia. Zhou et al29 observaron que tras seis meses de una angioplastia transluminal percutánea la frecuencia de serología positiva frente a CMV era de un 43% en los pacientes con arterias estenosadas, frente a un 8% de arterias estenosadas sin CMV (p < 0,002).
Estos estudios presentan las mismas dificultades para establecer conclusiones que los realizados para H. pylori. La mayoría cuentan con un escaso número de pacientes y de igual forma existe una falta de ajuste frente a posibles factores de confusión, aunque en este caso hay mayor número de diseños prospectivos. Además son pocos los estudios realizados en cardiopatía isquémica clásica, puesto que prioritariamente están realizados en receptores de trasplante cardíaco, en reestenosis coronaria y en aterosclerosis carotídea.
Mención aparte merecen los estudios en receptores de trasplante cardíaco. Desde hace tiempo se sabe que los receptores de un trasplante cardíaco desarrollan una aterosclerosis acelerada que, en muchas ocasiones, es la responsable de la morbimortalidad de estos enfermos30. En la población trasplantada el CMV está presente hasta en el 80% de los pacientes. En un estudio realizado en la universidad de Stanford31 se encontró que la aterosclerosis postrasplante era más frecuente y precoz en los pacientes con serología positiva frente a CMV. A los cinco años de la cirugía el 90% de los pacientes con serología inicial negativa frente a CMV mantenían las arterias coronarias sin lesiones ateroscleróticas, por sólo el 72% de los pacientes con serología inicial positiva. En los pacientes que fallecieron tras cinco años del trasplante se encontraron estenosis de más del 50% en las coronarias en un 7,6% de los pacientes CMV positivos, frente a un 0,8% en los negativos.
Estudios patológicos
La presencia de CMV ha sido demostrada en todo el árbol arterial, tanto en lesiones ateroscleróticas como en arterias normales32. No está claro si es más frecuente en las arterias lesionadas o en las sanas. Se cree, por tanto, que uno de los lugares de latencia del virus pueda ser la pared arterial. En un metaanálisis de 16 estudios que analizaban arterias de pacientes trasplantados se encontró CMV en un 47% de las arterias con arterioesclerosis frente a un 39% en las arterias sin lesiones, con una OR de 1,4 (95% CI: 1,0-1,9)17. Un inconveniente es que los resultados no son reproducibles con las diferentes técnicas de aislamiento, existiendo una importante variabilidad. En los estudios que utilizan la reacción en cadena de la polimerasa (PCR) se detectó la presencia de CMV en un 57% de las lesiones ateroscleróticas frente a un 36% en las arterias sanas con una OR de 2,5 (95% CI: 1,6-3,8)17. Aunque estos resultados son significativos, no aportan suficiente evidencia, puesto que las diferencias no son grandes y los CI son excesivamente amplios.
Se han propuesto diversos mecanismos que intentan explicar la producción de aterosclerosis. Parece que la infección por virus de la familia herpes facilita el acúmulo de colesterol en el interior de las células musculares lisas mediante un aumento de la captación de las LDL-ox. Asimismo, las infecciones víricas condicionan un estado procoagulante en el endotelio y el mecanismo más importante sería la exposición al torrente circulatorio del factor tisular, que es un poderoso coagulante33.
El papel del CMV en la reestenosis tras angioplastia parece tener una explicación patogénica en la expresión de genes víricos tempranos que se producen tras una infección abortiva de CMV. Dos de estos productos tempranos (IE72 e IE84) han sido relacionados con el proceso de proliferación celular. La proteína IE72 provoca un aumento en la expresión de receptores de LDL-ox, favoreciendo el acúmulo de colesterol en las células musculares lisas13. Se piensa que el IE84 se uniría al protooncogén p53 y lo inactivaría, alterando gravemente el ciclo celular. Impediría la finalización del ciclo celular, perpetuándose y provocando una inhibición de la apoptosis que induciría la proliferación de las células musculares lisas11. Se especula con que el daño producido por la dilatación mecánica de la pared arterial mediante angioplastia puede estimular la infección latente por el CMV.
Modelos animales
La evidencia del posible papel patogénico del CMV en la aterosclerosis proviene de los experimentos en animales realizados por Fabricant y Hajjar12,28. En los años cuarenta, Paterson y Cottral5 descubrieron una asociación entre la aterosclerosis y el virus de la enfermedad de Mareck. Posteriormente, Fabricant inyectó virus de la enfermedad de Mareck a pollos sanos y les sometió a una dieta rica en colesterol, escogiendo como controles a pollos no infectados pero alimentados con la dieta rica en lípidos. En las autopsias había lesiones aterosclerosas en toda la pared arterial de los pollos infectados, mientras que estas lesiones eran mínimas o inexistentes en los sanos. También observó que estas lesiones eran más extensas en aquellos pollos infectados y alimentados con la dieta rica en colesterol. La vacunación previa de los pollos contra el virus de la enfermedad de Mareck les protegía frente al acúmulo de lípidos en la pared arterial34.
Los experimentos de Hajjar en la década de los ochenta demostraron que la infección de pollos con el virus de la enfermedad de Mareck provocaba un mayor acúmulo de colesterol, ésteres de colesterol, triglicéridos y fosfolípidos en las paredes aórticas12 .
Ensayos de intervención terapéutica
No existen estudios bien diseñados que aporten evidencias del beneficio del tratamiento frente a la infección por CMV. Un estudio experimental en ratas en las que se realizaba un trasplante cardíaco sugiere que la administración de ganciclovir a ratas portadoras de CMV disminuye los efectos de la vasculopatía del trasplante35.
Algunos estudios realizados en receptores humanos de trasplante cardíaco infectado por CMV sugieren que la administración profiláctica de ganciclovir reduce la incidencia de enfermedad vascular relacionada con el virus36, pero hasta la fecha no hay trabajos que permitan establecer la eficacia de ganciclovir en la reducción de la incidencia de aterosclerosis.
Aterosclerosis y Chlamydia pneumoniae
Chlamydia pneumoniae es una bacteria gramnegativa, de crecimiento intracelular. Se transmite a través de las secreciones respiratorias y se cree que persiste en el interior de los macrófagos alveolares. Es el segundo patógeno causante de neumonías atípicas, responsable del 10% de los casos. La primoinfección por C. pneumoniae suele ocurrir en edades tempranas de la vida y las reinfecciones son extraordinariamente frecuentes. Aproximadamente el 50% de los mayores de 50 años tienen serología positiva frente a C. pneumoniae y su prevalencia se correlaciona con la edad, el nivel socioeconómico, el hábito tabáquico y las epidemias periódicas. Es mucho más frecuente en los varones37.
Es extraordinariamente difícil distinguir entre infecciones agudas o crónicas y, dentro de estas últimas, entre reagudizaciones y cronicidad. Esto es debido a que los métodos de detección no son fiables y a que las formas clínicas son superponibles. Existen diversos métodos de identificación en el laboratorio. El método de fijación de complemento no es útil, puesto que no diferencia entre las distintas especies de Chlamydia debido a que el lipopolisacárido de la cubierta es muy similar en todas ellas. Las técnicas de inmunofluorescencia son las más utilizadas en los estudios, pero son muy complejas y requieren experiencia a la hora de interpretarlas. Las técnicas mediante PCR no están muy extendidas y su sensibilidad no es muy buena. El cultivo es extraordinariamente difícil, requiere medios celulares y su sensibilidad es menor que las técnicas serológicas.
Otro problema añadido es la falta de unanimidad a la hora de definir los títulos de anticuerpos. Si bien existe consenso en que la infección aguda se caracteriza por un aumento en los títulos de IgM seguido más tarde de una pequeña elevación en los títulos de IgG o bien una seroconversión de al menos un aumento de cuatro veces los títulos de IgG, no está clara la distinción entre infección crónica persistente y reinfección. Se ha dado valor a la elevación persistente de IgA específica junto con aumento de IgG como marcador de infección crónica debido a que la vida media de la IgA es más corta que la de IgG38.
Estudios seroepidemiológicos
Los resultados de los estudios seroepidemiológicos muestran una asociación fuerte entre serología positiva frente a C. pneumoniae y aterosclerosis, con una OR > 217.La mayoría de ellos están realizados en cardiopatía isquémica clásica (angina de pecho, infarto de miocardio) y algunos en patología cerebrovascular. El trabajo de Saikku, publicado en 19882, marcó la pauta para el desarrollo de trabajos ulteriores. En este estudio, realizado en una población de 40 enfermos con infarto agudo de miocardio, en 30 con cardiopatía isquémica crónica y en 41 controles, cerca del 70% de los pacientes con infarto de miocardio tenía serología positiva frente a C. pneumoniae a títulos >1:128. El 50% de los que padecían cardiopatía isquémica crónica tenían niveles de IgG e IgA significativamente mayores que los controles. No se encontraron diferencias entre los pacientes con infarto y aquellos con angina de pecho. Sin embargo, no todos los trabajos muestran una asociación positiva a pesar de los títulos altos de anticuerpos frente a Chlamydia39.
Como ocurre con los trabajos realizados con CMV y Helicobacter, el principal inconveniente es que la mayoría son estudios caso/control y los escasos estudios prospectivos incluyeron pocos pacientes. De igual forma, no todos realizan ajuste frente a posibles factores de confusión.
La comparación entre los diferentes estudios se hace imposible. La utilización de distintos métodos de cuantificación de anticuerpos frente a Chlamydia (en la mayoría por inmunofluorescencia, aunque en otros se utiliza detección de inmunocomplejos) y la falta de acuerdo a la hora de definir infección crónica por criterios serológicos son los principales inconvenientes de dichos estudios. En todos ellos se utilizan distintos puntos de corte.
Estudios patológicos
Existen numerosas evidencias patológicas que relacionan la infección por C. pneumoniae con la arterioesclerosis de cualquier localización.Thom et al40 encontraron correlación entre los niveles de anticuerpos y la severidad de las lesiones angiográficas. En diferentes estudios de muestras obtenidas por necropsia se ha encontrado la bacteria en las placas de ateroma, pero también en las células musculares lisas y en las células espumosas, mientras que no se evidenciaba en las paredes arteriales sanas17. La mayoría de estos estudios identifican a la Chlamydia por técnicas de inmunohistoquímica o PCR. También ha sido posible detectar la bacteria en muestras in vivo en carótidas, aorta abdominal, arterias periféricas y, recientemente, en válvulas aórticas no reumáticas41,42. En algunos estudios se ha llegado a aislar la bacteria mediante cultivo en líneas celulares.
En trece estudios publicados que demuestran la presencia de C. pneumoniae en muestras patológicas en función de la presencia de ADN, antígenos o cuerpos elementales, la infección local se confirmó en un 52% de las lesiones ateromatosas por sólo un 5% de los controles con arterias sanas17. Aunque ello no significa que en las lesiones haya bacterias viables, en ocasiones se ha conseguido su cultivo en muestras patológicas.
Los mecanismos propuestos para explicar esta asociación han sido diversos. Algunos trabajos encuentran que aquellos con títulos elevados de anticuerpos frente a Chlamydia presentan concentraciones plasmáticas elevadas de proteína C reactiva y de fibrinógeno43,44. Asimismo, la infección podría desencadenar la liberación de mediadores químicos por parte de los macrófagos (interleucina 1, interferón *, FNT-*) que estimulan la respuesta inflamatoria.
Al igual que con H. pylori, el daño endotelial en la infección por Chlamydia puede desencadenarse por una reacción cruzada frente a determinantes antigénicos similares, en este caso las HSP6045.
A pesar de estas evidencias, no es posible establecer si la infección por C. pneumoniae desencadena el proceso aterosclerótico o, por el contrario, acelera este proceso una vez iniciado por los factores de riesgo ateroscleróticos clásicos.
Modelos animales
Existen dos modelos animales experimentales de infecciónpor Chlamydia muy interesantes. El primero se desarrolló en ratones modificados genéticamente para ser deficientes en apolipoproteína E, que condiciona una predisposición a la arterioesclerosis46. Se inoculó C. pneumoniae a ratones deficientes y a ratones normales. En los ratones deficientes se detectó una infección persistente de Chlamydia en las placas de ateroma con presencia de la bacteria en muestras de las placas, mientras que en los ratones sin el defecto genético se evidenciaron infecciones transitorias.
Recientemente se ha desarrollado otro modelo de aterosclerosis en conejos47. Se inoculó C. pneumoniae por vía intranasal a conejos sanos y a otros se les inoculaba placebo. Diez de los once conejos inoculados con la bacteria demostraron infección por medio de métodos serológicos. Varios de estos animales tenían evidencias de cambios ateroscleróticos en sus arterias y se aisló C. pneumoniae en dos aortas, observándose cuerpos elementales, y en un caso se aisló por cultivo. En el grupo control no hubo evidencia de infección ni de cambios ateroscleróticos en las paredes arteriales. Se especuló que la infección por Chlamydia se adquiriría en edades tempranas en forma de infección respiratoria y la bacteria quedaría acantonada en el interior de los macrófagos alveolares. Posteriormente éstos migrarían por el torrente sanguíneo y se incorporarían en aquellos lugares en los que el endotelio hubiese sufrido daño por cualquier causa, con lo que se internarían en el intersticio arterial y ahí se perpetuarían. Una reinfección tardía estimularía la respuesta inmune de linfocitos y macrófagos, reactivándose la respuesta inflamatoria en la pared arterial creando una placa de ateroma más vulnerable.
Utilizando este modelo animal hay un estudio de intervención terapéutica que apoya un posible papel protector de la azitromicina en la prevención de estas lesiones48. Se inoculó Chlamydia por vía intranasal a conejos a los que se les administraba una dieta rica en colesterol. A una pequeña muestra aleatoria de éstos se les administró durante siete semanas azitromicina. Tras este período se sacrificó a los animales y se observó que la población tratada con azitromicina mostraba un menor grosor de la íntima arterial que los no tratados (0,20 frente a 0,55 mm, respectivamente). Sin embargo, otros trabajos experimentales no han podido demostrar esta asociación. Así, Moazed et al49 no consiguieron reproducir lesiones ateroscleróticas tras la inoculación intranasal de Chlamydia.
Ensayos de intervención terapéutica
Hasta la fecha sólo existen dos grandes estudios de intervención terapéutica en humanos, aunque actualmente hay varios trabajos en desarrollo que intentan demostrar un efecto beneficioso del tratamiento con antibióticos. El primer estudio de Gurfinkel et al50 (estudio ROXIS) evaluó el posible papel protector de la roxitromicina en la prevención de nuevos episodios isquémicos. Seleccionaron 202 pacientes (la mayoría varones) que habían sufrido un infarto agudo de miocardio o un episodio de angina inestable. Se distribuyeron aleatoriamente en dos grupos, para recibir roxitromicina, 150 mg dos veces al día durante treinta días, o placebo. El período de seguimiento del estudio fue de seis meses y como eventos finales se estimaron la muerte por cardiopatía isquémica, nuevo episodio de infarto o isquemia recurrente. El análisis de los eventos por separado demostró una cierta tendencia beneficiosa, pero no estadísticamente significativa. Cuando se analizaron los tres eventos finales juntos se obtuvo una tasa significativamente menor de sucesos en el grupo activo que en el placebo (1% frente a 10%, p = 0,032).
El otro gran estudio realizado por Gupta et al51 incluyó a 213 varones que habían sufrido un infarto de miocardio. Los enfermos fueron divididos en tres grupos según los títulos de anticuerpos frente a C. pneumoniae. Un grupo con serología negativa, otro con títulos entre 1/8 y 1/32 y el último con títulos superiores o igual a 1/64. A este último grupo se les subdividió aleatoriamente en tres subgrupos: unos recibieron azitromicina 500 mg/día durante tres días, otros recibieron dos tandas de azitromicina de tres días, separados entre sí tres meses, y el último grupo recibió placebo. El período de seguimiento fue de 18 meses. El primer objetivo del trabajo era comparar la reducción de los títulos de anticuerpos. Los objetivos secundarios eran analizar la disminución de episodios coronarios: muerte de causa cardíaca, infarto de miocardio no fatal, angina inestable e infarto no-Q o angina inestable susceptible de coronariografía. Después de seis meses un 43% de los que recibieron antibiótico redujeron sus títulos de anticuerpos hasta 1/16 frente a un 10% de los que recibieron placebo (p < 0,02). Analizando los objetivos secundarios encontraron que en el grupo tratado con azitromicina el riesgo de acontecimientos clínicos era cuatro veces inferior a los no tratados (p = 0,03).
Aunque estos estudios aportan cierta evidencia en favor del protagonismo de la Chlamydia en el desarrollo de aterosclerosis, no están exentos de críticas metodológicas. Ambos se realizaron con escaso número de pacientes y no se tuvo en cuenta el posible efecto beneficioso del tratamiento antibiótico frente a microorganismos diferentes de C. pneumoniae. Tampoco se consideró el que parte del beneficio podría derivarse del efecto antiinflamatorio que tienen los macrólidos. Además en ninguno de los estudios se hizo una monitorización estrecha del cumplimiento terapéutico. En el trabajo de Gurfinkel no se tiene en cuenta los títulos de anticuerpos, se aplica el tratamiento antibiótico independientemente del grado de infección. Por el contrario, en el trabajo de Gupta, aunque se consideraron los títulos de anticuerpos, no se hizo una distinción entre infección pasada o reciente.
Estos inconvenientes hace que sean necesarios más estudios y en mayor escala que permitan establecer un beneficio claro de la intervención terapéutica, estratificar a los pacientes según el riesgo para desarrollar aterosclerosis y poder seleccionar aquellos que se beneficiarían más del tratamiento antibiótico. Los estudios a gran escala que se encuentran actualmente en desarrollo (Academic, Wizard, Stamina, Marble) podrían aclarar estos aspectos todavía oscuros.
Conclusiones
Es posible que los procesos infecciosos puedan intervenir en la patogenia del fenómeno aterosclerótico, aunque existen todavía muchas sombras que deben ser aclaradas. La hipótesis de respuesta a la lesión desarrollada por Ross permite apreciar la aterosclerosis como un fenómeno inflamatorio. Esta hipótesis, hoy globalmente aceptada, nos ofrece posibles vías de actuación de agentes infecciosos perpetuando o provocando el fenómeno inflamatorio que va a desembocar en la placa de ateroma. Hasta la fecha las mayores evidencias favorecen al CMV y Chlamydiapneumoniae. Los distintos argumentos en favor y en contra quedan reflejados en la tabla 3. Por el contrario, hay escasas evidencias que apoyen el papel de Helicobacter pylori en el desarrollo de aterosclerosis.
El CMV parece más relacionado con la aterosclerosis desarrollada tras el trasplante cardíaco, así como con el fenómeno de reestenosis tras una angioplastia coronaria.
Los datos más consistentes corresponden a la asociación con C. pneumoniae, aunque hacen falta muchos más estudios fundamentalmente prospectivos y con mayor número de personas con ajuste frente a las múltiples variables de confusión.
Por último, los estudios de intervención terapéutica nos ofrecen una herramienta más en la lucha contra la aterosclerosis, aunque queda mucho camino por delante hasta demostrar el hipotético beneficio del uso de antibióticos en la aterosclerosis. No sólo son necesarios más estudios y mejor diseñados que demuestren la regresión o menor aparición de placas de ateroma, sino que debe considerarse el problema de las resistencias antibióticas que resultaría del uso indiscriminado de estos fármacos.
Un mejor conocimiento de la asociación entre aterosclerosis e infección puede ayudarnos a conseguir disminuir la morbimortalidad asociada a esta enfermedad, que es responsable del 20% de las muertes en todo el mundo.