







Las microangiopatías trombóticas (MAT) son un grupo de entidades que se caracterizan por presentar una anemia hemolítica microangiopática (con los típicos esquistocitos en el frotis de sangre periférica), trombocitopenia y afectación de órganos de intensidad variable. La púrpura trombocitopénica trombótica y el síndrome urémico hemolítico son las formas más importantes de MAT, y sin el tratamiento adecuado se asocian a una elevada morbimortalidad. En los últimos años se han producido avances notables en el conocimiento de la fisiopatología de las MAT. Estos avances nos han permitido pasar de un diagnóstico sindrómico con un tratamiento similar en todos los casos, a buscar un diagnóstico etiológico y un tratamiento acorde a la etiología que ha conllevado una mejoría en el pronóstico de los pacientes. Este documento pretende resumir el estado actual del conocimiento de la fisiopatología y las opciones terapéuticas disponibles, y también presentar a los profesionales que tratan a este tipo de pacientes una aproximación diagnóstica y terapéutica práctica.
Thrombotic microangiopathies (TMA) are disorders defined by the presence of a microangiopathic hemolytic anemia (with the characteristic hallmark of schistocytes in the peripheral blood smear), thrombocytopenia and organ malfunction of variable intensity. Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome are the most important forms of TMA and, without the adequate treatment, they are associated with high morbimortality. In recent years, significant advances in the knowledge of the pathophysiology of TMA have occurred. Those advances have allowed us to move from a syndromic diagnosis with a similar treatment to all entities to the search of etiologic diagnosis which would lead to a specific treatment, finally leading to a better outcome of the patient. This document pretends to summarize the current status of knowledge of the pathophysiology of TMA and the therapeutic options available, and to offer a diagnostic and therapeutic practical tool to the professionals caring for the patients.
Probablemente, en los últimos años, las microangiopatías trombóticas (MAT) han sido uno de los campos de la medicina donde se han producido los cambios más importantes en nuestros conocimientos tanto en su fisiopatología como en su tratamiento. En efecto, en casi 90 años hemos pasado desde la primera descripción de Eli Moschcowitz en 1925 de una enfermedad hasta entonces no descrita y que él atribuyó a «un potente veneno con efectos aglutinantes y hemolíticos»1, a poder precisar la mutación responsable de las formas congénitas de la púrpura trombocitopénica trombótica (PTT) o a estar desarrollando péptidos inhibidores de la activación de C32 para tratar la hiperactivación del complemento observada en el síndrome urémico hemolítico atípico (SUHa).
Debido a estos rápidos avances en el conocimiento y tratamiento de las MAT acontecidos recientemente, el Grupo Español de Aféresis de la Sociedad Española de Hematología y Hemoterapia y de la Sociedad Española de Transfusión Sanguínea y Terapia Celular decidió constituir un grupo de trabajo para publicar una guía donde se resumiera el estado actual del diagnóstico y tratamiento de este tipo de entidades.
Los autores de la guía confían en que el trabajo realizado sea útil para los especialistas que tratan este tipo de entidades y les ayude en el complejo proceso de diagnosticar y tratar a los pacientes afectados de una MAT.
Definición y epidemiologíaLas MAT son un conjunto de procesos que cursan con una alteración del endotelio vascular y presentan hallazgos característicos en el frotis de sangre periférica de una anemia hemolítica microangiopática (esquistocitos), datos de laboratorio característicos (elevación de reticulocitos y lactato deshidrogenasa [LDH]) y trombocitopenia de intensidad variable.
La lista de entidades asociadas a la aparición de una MAT es amplia (tabla 1). Las entidades más importantes dentro de las MAT son la PTT en sus variedades congénita y adquirida (autoinmunitaria), el SUH típico asociado a infección por Escherichia coli (E. coli) productora de toxina Shiga (SUH asociado a diarrea o STEC-SUH) y el SUHa. Tanto la PTT como las diferentes variedades de SUH son entidades muy poco frecuentes. De la PTT congénita (síndrome de Upshaw-Schulman) se han publicado algo más de 100 casos en la literatura médica, y su incidencia y prevalencia exacta se desconocen, aunque probablemente estén subestimadas. Es más frecuente en la infancia y la adolescencia, pero puede aparecer a cualquier edad. La PTT autoinmunitaria tiene una incidencia de unos 4-6 casos nuevos por millón de habitantes y año en EE. UU.3 y de 6 casos por millón de habitantes y año en el Reino Unido4, no existiendo datos fiables en España. Tiene un pico máximo de incidencia en la cuarta década de la vida, con un discreto predominio de mujeres (3:2) y entre personas de raza negra. La tabla 2 muestra la incidencia de la PTT recogida en el registro japonés de MAT5.
Clasificación de las microangiopatías trombóticas
| Microangiopatías trombóticas primarias | |
|---|---|
| Entidad | Fisiopatología |
| SUH asociado a infección | Escherichia coli cepa O157:H7 y otras cepas no O157:H7, Shigella dysenteriae tipo i, Staphylococcus pneumoniae (neuraminidasa) |
| SUHa | Alteraciones genéticas del complemento: mutaciones en FHC, PCM, FIC, THBD, FBC y C3. Alteraciones inmunitarias del complemento: anticuerpos anti-FHC |
| PTT idiopática | Alteraciones inmunitarias de ADAMTS13 (actividad<5-10%) con anticuerpos inhibidores |
| PTT congénita (síndrome de Upshaw-Schulman) | Alteraciones genéticas de ADAMTS13 (actividad<5-10%) sin la presencia de anticuerpos inhibidores |
| Microangiopatías trombóticas secundarias | |
|---|---|
| Tipo de desencadenante | |
| Fármacos | Quinina, mitomicina C, gemcitabina, cisplatino, interferón, inhibidores de VEGF y tirosín cinasa, ticlopidina, clopidogrel, inhibidores de la calcineurina (ciclosporina, tacrolimus), sirolimus, valaciclovir, anticonceptivos orales |
| Conectivopatías | LES; síndrome antifosfolípido; esclerodermia |
| Gestación | HELLP, preeclampsia |
| Otras | Infección por VIH; glomerulopatías; hipertensión arterial maligna; H1N1 (influenza A); neoplasias; aciduria metilmalónica con homocistinuria; TPH, TOS |
FBC: factor B del complemento; FHC: factor H del complemento; FIC: factor I del complemento; HELLP: síndrome anemia hemolítica, elevación de las transaminasas y trombocitopenia; LES: lupus eritematoso sistémico; PCM: proteína cofactor de membrana; PTT: púrpura trombocitopénica trombótica; SUH: síndrome urémico hemolítico; SUHa: síndrome urémico hemolítico atípico; THBD: trombomodulina; TOS: trasplante de órgano sólido; TPH: trasplante de progenitores hematopoyéticos; VEGF: vascular endothelial growth factor («factor de crecimiento vascular endotelial»); VIH: virus de la inmunodeficiencia humana.
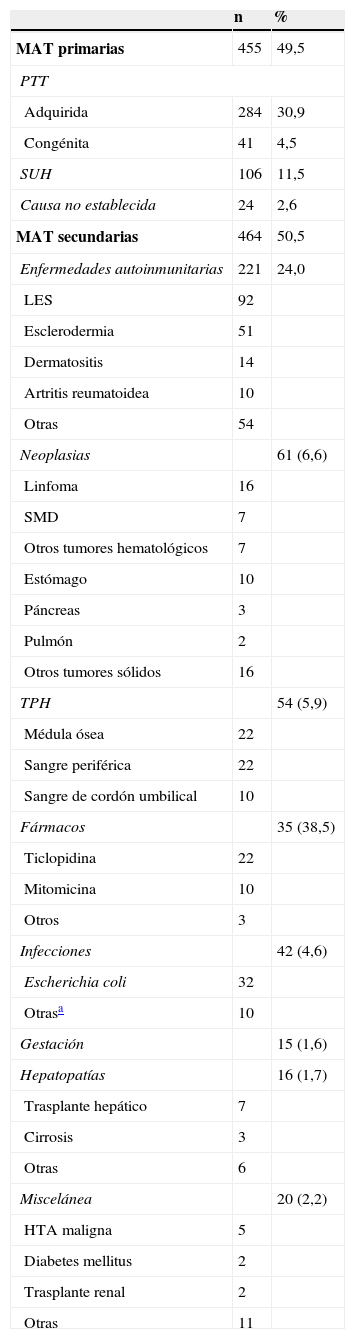
Frecuencia de las microangiopatías trombóticas recogidas en el registro japonés (n=919)
| n | % | |
|---|---|---|
| MAT primarias | 455 | 49,5 |
| PTT | ||
| Adquirida | 284 | 30,9 |
| Congénita | 41 | 4,5 |
| SUH | 106 | 11,5 |
| Causa no establecida | 24 | 2,6 |
| MAT secundarias | 464 | 50,5 |
| Enfermedades autoinmunitarias | 221 | 24,0 |
| LES | 92 | |
| Esclerodermia | 51 | |
| Dermatositis | 14 | |
| Artritis reumatoidea | 10 | |
| Otras | 54 | |
| Neoplasias | 61 (6,6) | |
| Linfoma | 16 | |
| SMD | 7 | |
| Otros tumores hematológicos | 7 | |
| Estómago | 10 | |
| Páncreas | 3 | |
| Pulmón | 2 | |
| Otros tumores sólidos | 16 | |
| TPH | 54 (5,9) | |
| Médula ósea | 22 | |
| Sangre periférica | 22 | |
| Sangre de cordón umbilical | 10 | |
| Fármacos | 35 (38,5) | |
| Ticlopidina | 22 | |
| Mitomicina | 10 | |
| Otros | 3 | |
| Infecciones | 42 (4,6) | |
| Escherichia coli | 32 | |
| Otrasa | 10 | |
| Gestación | 15 (1,6) | |
| Hepatopatías | 16 (1,7) | |
| Trasplante hepático | 7 | |
| Cirrosis | 3 | |
| Otras | 6 | |
| Miscelánea | 20 (2,2) | |
| HTA maligna | 5 | |
| Diabetes mellitus | 2 | |
| Trasplante renal | 2 | |
| Otras | 11 | |
HTA: hipertensión arterial; LES: lupus eritematoso sistémico; MAT: microangiopatías trombóticas; SMD: síndromes mielodisplásicos; TPH: trasplante de progenitores hematopoyéticos; PTT: púrpura trombocitopénica trombótica; SUH: síndrome urémico hemolítico.
La incidencia anual de STEC-HUS es de cerca de 2 casos por 100.000 habitantes en adultos y de 6 casos por 100.000 en niños menores de 5 años. El SUHa supone aproximadamente el 5% de los SUH y puede aparecer a cualquier edad, con especial incidencia en niños y adultos jóvenes. Se considera una enfermedad ultrarrara, con una incidencia en EE. UU. de 1-2 casos/millón de habitantes en personas de hasta 18 años y sin datos definitivos en la población adulta6, mientras que en Europa se estima su incidencia en 0,11 casos/millón de habitantes. Es de prever, sin embargo, que el mejor conocimiento de la fisiopatología de estos procesos se acompañe de un incremento en el número de pacientes diagnosticados en el futuro inmediato (tabla 3).
Pruebas de laboratorio para el diagnóstico diferencial de una microangiopatía trombótica
| Pruebas cuyos resultados se obtienen en general de urgencia |
| Hemograma completo |
| Reticulocitos |
| Examen del frotis de sangre periférica |
| Prueba de antiglobulina directa |
| Pruebas de coagulación (tiempo de protrombina, tiempo de tromboplastina parcial activado, fibrinógeno y productos de degradación de la fibrina) |
| Perfil bioquímico (creatinina, bilirrubina total e indirecta, ASAT, ALAT y LDH) |
| Pruebas cuyos resultados se obtienen en general en días |
| Actividad e inhibidor de ADAMTS13 |
| Haptoglobina |
| Serología para los virus de la inmunodeficiencia humana y hepatitis B y C |
| Cribado de enfermedades autoinmunitarias: anticuerpos antinucleares, anti-ADN, anti-antígeno nuclear extraíble |
| Proteínas y actividad del complemento (C3, C4, CH50, factor I, factor H, factor B) |
| Niveles plasmáticos de vitamina B12 |
| Homocisteína en plasma y niveles plasmáticos y urinarios de ácido metilmalónico |
| Detección toxina Shiga en heces o en cultivo de heces |
| Pruebas cuyos resultados se obtienen en general en semanas |
| Estudio de genes de proteínas del complemento |
| Anticuerpos anti-factor H |
ALAT: alanino aminotransferasa; ASAT: aspartato aminotransferasa; LDH: lactato deshidrogenasa.
El desarrollo de una MAT puede aparecer asociado a diferentes trastornos o fármacos; son las denominadas MAT secundarias (tabla 2)5.
Clasificación de las microangiopatías trombóticas. Etiología y fisiopatologíaPúrpura trombocitopénica trombóticaTanto la forma congénita como la autoinmune o adquirida aparecen como consecuencia de un déficit o disfunción de una proteína denominada a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13, «una desintegrina y metaloproteasa con motivo trombospondina tipo1, 13»), responsable de la escisión del factor von Willebrand (FvW). Identificada en 19967,8, no fue hasta 2001 cuando fue denominada ADAMTS13, al determinar su estructura y constatar su homología con otros miembros de la familia de metaloproteasas ADAMTS9.
En condiciones fisiológicas, el FvW es liberado por las células endoteliales y queda adherido a la superficie de la célula, donde la metaloproteasa ADAMTS13 actúa a nivel del dominio A2 del FvW escindiendo los multímeros ultragrandes que presentan una gran afinidad por las plaquetas. A continuación, el FvW es liberado a la circulación sanguínea con una conformación que no permite su interacción con las plaquetas. La actividad de ADAMTS13 es dependiente de la presencia de los cationes divalentes calcio y cinc.
El defecto congénito o adquirido de ADAMTS13 hace que la molécula de FvW conserve los multímeros ultragrandes10,11, que provocarán la adhesión de plaquetas a través del complejo glucoproteico GPIb-V-IX. Ello da lugar a la formación de trombos plaquetarios sobre el endotelio alargados en la dirección del flujo, que en un momento dado se romperán y acabarán provocando la aparición de complicaciones clínicas con la fragmentación de los hematíes por el flujo turbulento, la disminución de plaquetas por el consumo en los trombos y el aumento de la LDH sérica con un desproporcionado aumento de la isoenzima 5 proveniente del músculo y el hígado. Ello evidencia que la isquemia tisular sistémica desempeña un papel significativo en el aumento de LDH12.
Las raras formas congénitas están ocasionadas por mutaciones homocigotas o doble heterocigotas del gen ADAMTS13, de las que se han descrito más de 100 tipos diferentes9,13, mientras que las formas adquiridas lo son por la acción de autoanticuerpos (IgG, IgA o IgM) dirigidos contra esta enzima que ocasionan que su actividad en plasma descienda a valores por debajo del 5-10% de la normalidad. Estos anticuerpos pueden aparecer sin causa desencadenante (PTT idiopática) o asociados a la presencia de factores precipitantes, como conectivopatías, cáncer, gestación y ciertos fármacos (MAT secundarias). Hay que destacar el hecho de que hasta en un 5% de la población sana se han descrito anticuerpos IgG anti-ADAMTS13 de bajo título que reconocen epítopos similares a los anticuerpos identificados en pacientes14. En la mayor parte de los pacientes que alcanzan remisión, los anticuerpos desaparecen y se normalizan los valores de ADAMTS13; sin embargo, algunos presentan valores persistentemente bajos de ADAMTS13 a pesar de alcanzar una remisión clínica, lo que demuestra la necesidad de algún suceso adicional necesario para el desarrollo de un episodio de PTT. Los nucleosomas circulantes encontrados durante las fases agudas de la enfermedad y que desaparecen durante las fases de remisión pueden ser reflejo de este segundo suceso15. Esos nucleosomas podrían, en parte, derivar de neutrófilos activados atrapados en la matriz extracelular durante algún proceso infeccioso previo habitualmente leve. Por último, algunos autores han demostrado que otro factor que podría incrementar el riesgo de desarrollar autoinmunidad frente a ADAMTS13 sería la presencia de ciertos alelos HLA-DR, como el B1*1116,17.
Síndrome urémico hemolíticoComo en la PTT, 2 son las variantes de SUH identificadas. La forma más frecuente, normalmente de buen pronóstico, va precedida de un cuadro de diarrea hemorrágica y aparece tras una infección, habitualmente intestinal por cepas de E. coli productora de toxina Shiga (STEC-SUH), aunque también pueden ocasionar un SUH sin diarrea asociada a ciertas cepas de Streptococcus pneumoniae. La segunda variante supone el 5-10% de todos los SUH, se asocia a alteraciones congénitas (o más rara vez adquiridas) de la vía alternativa del complemento y se denomina SUHa.
Síndrome urémico hemolítico asociado a infecciónEs más común en niños menores de 5-6 años y en la variante más frecuente su aparición se debe a la acción de las toxinas liberadas por ciertos microorganismos, especialmente E. coli O157:H7 y el serotipo 1 de Shigella dysenteriae (S. dysenteriae), y su curso clínico es normalmente autolimitado. Los gérmenes suelen penetrar en el organismo por vía digestiva a través de alimentos contaminados, proliferando en la luz intestinal y adhiriéndose a las células epiteliales de la mucosa del colon, donde liberan toxinas que lesionan el tejido subyacente y la vascularización, produciendo una diarrea hemorrágica. Atravesada la barrera intestinal, las toxinas son transportadas por neutrófilos, monocitos y plaquetas hasta la microcirculación renal, donde estimulan la liberación de citocinas como el tumor necrosis factor α (TNF-α, «factor de necrosis tumoral α»), la interleucina 1 (IL-1) y la IL-6 a partir de los monocitos y las células del glomérulo y del epitelio renal. Esto facilita su internalización en las células endoteliales de los capilares glomerulares, células mesangiales y células epiteliales glomerulares y tubulares. Una vez dentro de las células, la subunidad A de la toxina Shiga inhibe la subunidad 28S del ribosoma y, por tanto, la síntesis proteica, lo que conduce a la apoptosis y la lesión del endotelio renal. Esta lesión endotelial facilita la aparición de un estado protrombótico local con un incremento en la adhesión plaquetar y la aparición de microtrombos ricos en fibrina18.
Solamente del 5-15% de los pacientes que presentan una gastroenteritis asociada a STEC desarrollan un SUH, lo que evidencia que existen otros factores, hasta ahora desconocidos, que también desempeñan un papel en la aparición de estos cuadros. Entre estos se pueden encontrar mutaciones o polimorfismos de diferentes genes, incluyendo el receptor de la toxina Shiga, el TNF-α y la IL-1.
Síndrome urémico hemolítico atípicoOcasionalmente, los pacientes pueden presentar hallazgos típicos de SUH, pero sin evidencia de infección bacteriana previa. Es el denominado SUHa y su pronóstico es mucho peor que el del SUH clásico, con una mortalidad durante el primer episodio del 10-15%, y con un riesgo de desarrollar enfermedad renal terminal de hasta el 50% de los casos. Desde el punto de vista fisiopatológico existe una clara relación entre el SUHa y la presencia de defectos en la regulación de la vía alternativa del complemento. Las alteraciones descritas conducen a una activación excesiva de la vía alternativa del complemento que induce una lesión de las células endoteliales, activación plaquetaria y leucocitaria, y la aparición de trombosis en la microcirculación19,20.
El sistema del complemento está formado por un grupo heterogéneo de proteínas unidas a membrana o circulantes en plasma que sirven de protección frente a diferentes microorganismos. Tres son las vías de activación del complemento (clásica, de la lectina y alternativa) que producen complejos de proteasas, denominados convertasas C3 y C5, que escinden C3 y C5, respectivamente, y que originan la activación del complejo de ataque a la membrana, responsable de la lisis celular. La vía alternativa se activa de manera espontánea en el plasma por la hidrólisis de C3, que a su vez es la responsable del depósito de C3b en prácticamente todas las superficies expuestas al plasma. En la superficie bacteriana, C3b induce la fagocitosis y posterior opsonización bacteriana por los neutrófilos y macrófagos. De no existir una autorregulación, cualquier estímulo inicial ocasionaría una activación permanente del complemento que acabaría dañando incluso las células autógenas del paciente. En las células del huésped, esta activación excesiva en cascada es controlada por la presencia de reguladores tanto unidos a la membrana celular como circulantes21. Antígenos exógenos o células propias dañadas que carecen o no pueden unirse a estos reguladores son atacadas y eliminadas por el complemento activado.
Lo que existe en el SUHa es un desequilibrio de origen genético entre los factores activadores y reguladores de la actividad del complemento, que ocasiona una hiperactivación de la vía alternativa y que son los responsables de la aparición del cuadro clínico. Así, más del 50% de los pacientes con SUHa presentan mutaciones en FHC, FIC y PCM, que codifican las proteínas reguladoras del complemento factor H (FHC), factor I (FIC) y la proteína del cofactor de membrana (PCM). En un 3-5% de los pacientes se han encontrado genes híbridos derivados de un cruzamiento desigual entre FHC y FHCR1. Estos defectos genéticos ocasionan una disminución de la actividad reguladora del complemento sobre la superficie celular. También se han descrito anticuerpos inhibitorios en el 5-10% de los pacientes. Estos anticuerpos van casi siempre dirigidos contra la porción C-terminal de FHC, alterando, también, la regulación del complemento en la superficie celular. Además, se ha comprobado que el desarrollo de estos anticuerpos tiene una importante predisposición genética asociada a la deleción del gen FHCR1.
Mutaciones con ganancia de función en proteínas clave de esta vía alternativa como las que implican al factor C3 y al factor B (FBC) se han descrito en el 10 y el 1-2% de los pacientes, respectivamente. Cerca del 3% de los pacientes presentan mutaciones en genes que codifican la acción de la trombomodulina, glucoproteína de membrana con actividad procoagulante y que modula la activación del complemento en la superficie celular. También se han descrito casos de penetrancia incompleta de SUHa en pacientes con diferentes mutaciones, lo que demuestra que se necesitan factores ambientales o genéticos adicionales para que se desarrollen las manifestaciones clínicas de la enfermedad. Por último, diferentes hipótesis han intentado explicar por qué hay una tendencia a una mayor afectación de ciertos lechos vasculares. Así, la elevada frecuencia de lesión renal observada en estos pacientes podría deberse a la estructura fenestrada del glomérulo, que hace que la matriz subendotelial esté expuesta de manera continuada a las proteínas circulantes22.
Microangiopatías trombóticas secundariasComo ya se ha comentado previamente, además de las alteraciones en la regulación de los factores del complemento o el déficit de ADAMTS13 de origen autoinmunitario o congénito, la MAT puede aparecer asociada a diferentes factores precipitantes como neoplasias, fármacos, infecciones, enfermedades del tejido conectivo, etc. Estas MAT secundarias pueden asociarse a la presencia de anticuerpos anti-ADAMTS13 y/o a valores bajos (<10%) de esta proteína, y, de manera creciente, se están identificando alteraciones en la actividad del complemento en muchos de estos pacientes, especialmente en aquellos casos asociados a la gestación o a los trasplantes de progenitores hematopoyéticos (TPH) (tabla 2).
Manifestaciones clínicas de las microangiopatías trombóticasSi bien las MAT primarias son procesos sistémicos, la afectación de los órganos diana varía en frecuencia e intensidad, aunque a veces el solapamiento hace difícil su diagnóstico.
Púrpura trombocitopénica trombótica adquiridaPresenta habitualmente un curso clínico agudo, con tendencia a la recaída y asociación ocasional de otros trastornos autoinmunitarios. La descripción clínica clásica de la PTT ha sido la péntada de: anemia hemolítica microangiopática, trombocitopenia, fiebre, disfunción renal y alteraciones neurológicas23, pero no todos los pacientes la presentan. En un estudio reciente, al inicio de la enfermedad, la hemoglobina y las plaquetas estaban por debajo de 10g/dl y de 20×109/l en el 81 y el 73% de los casos, respectivamente, y la LDH se elevaba 4,5 veces el máximo de la normalidad. Desde el punto de vista clínico, había alteraciones neurológicas en el 63%de los episodios, diátesis hemorrágica en el 66% y fiebre en el 24%24.
Las manifestaciones neurológicas son típicamente focales, fluctuantes y recurrentes. Pueden existir confusión, cefalea, parestesias, disartria, afasia, alteración de la personalidad, deterioro cognitivo, accidentes isquémicos transitorios o cambios del nivel de consciencia, incluyendo el coma, que es un signo de mal pronóstico. La insuficiencia renal aparece en el 10-75% de los pacientes y normalmente es leve, siendo excepcional el desarrollo de fracaso renal agudo. La afectación cardiaca en pacientes con PTT suele ser infradiagnosticada y es una causa frecuente de mortalidad temprana25,26. La isquemia intestinal puede provocar dolor abdominal, náuseas y vómitos. A pesar de la trombocitopenia grave, las manifestaciones hemorrágicas importantes no son frecuentes.
El curso natural de la PTT sin tratamiento lleva a la muerte al 90% de los pacientes, a menudo antes de las primeras 24h, y se debe principalmente a episodios isquémicos (accidentes vasculares cerebrales, accidentes isquémicos transitorios, infarto de miocardio y arritmia). Un tercio de los supervivientes presentan una o más recaídas. Una parte considerable de los supervivientes ven afectada su calidad de vida, con depresión, trastornos de ansiedad y déficits neurocognitivos27.
Aunque más raramente, la PTT idiopática adquirida también se puede presentar en la población pediátrica (<18 años), siendo sus manifestaciones clínicas similares a las del adulto28,29.
Púrpura trombocitopénica trombótica congénita (síndrome de Upshaw-Schulman)Los pacientes con PTT congénita presentan de forma constante una actividad de ADAMTS13 muy disminuida. Independientemente de la edad al inicio, la mayoría de los pacientes presentan un curso crónico recurrente, como consecuencia de la exposición a alguno de los factores de riesgo mencionados, con un patrón clínico que suele repetirse en los siguientes episodios.
Se ha publicado una correlación genotipo-fenotipo, de modo que los pacientes con mutaciones de ADAMTS13 que conllevan una menor actividad basal comienzan más precozmente, con frecuencia requieren tratamiento con plasma, presentan más recurrencias y necesitan profilaxis secundaria con plasma30, aunque otros autores no han encontrado esto en su grupo de pacientes31. El 33-50% de los pacientes experimentan el primer episodio antes de los 5 años de edad, y en los neonatos puede enmascararse con una ictericia neonatal grave, siendo la clave diagnóstica la presencia de esquistocitos30. Clínicamente es similar a las formas adquiridas, aunque la afectación renal, a diferencia de las anteriores, puede ser una de las manifestaciones predominantes. Presenta alteraciones neurológicas (hemiparesia, hemiplejía o convulsiones) en el 35% de los casos.
Las formas que comienzan en la edad adulta suelen aparecer desencadenadas también por los factores de riesgo mencionados. En mujeres, el embarazo es un factor precipitante típico para el inicio13. Hay casos que no comienzan hasta los 50-60 años, en forma de accidentes isquémicos transitorios, accidentes cerebrovasculares aislados o enfermedad renal, en ocasiones sin microangiopatía concurrente. También se han publicado casos asintomáticos y otros con trombocitopenia aislada32.
Síndrome urémico hemolítico típicoEs la modalidad más frecuente de SUH en la infancia y es causa frecuente de insuficiencia renal aguda en esa edad. Debido a su origen infeccioso suele cursar en episodios agudos únicos, y, a diferencia de la PTT, puede ser endémico o epidémico. En el 80% de los casos derivados de infección por E. coli O157: H7 suele ir precedido por una diarrea sanguinolenta, a menudo muy dolorosa. Entre una y 2 semanas después aparece el cuadro de MAT y la insuficiencia renal, habitualmente prerrenal. Puede haber casos con diagnóstico tardío, con plaquetas normales y solo anemia residual33.
Aunque suele haber una recuperación completa en el episodio agudo, el pronóstico a largo plazo no siempre es bueno. La muerte o la insuficiencia renal crónica (IRC) ocurren en aproximadamente el 12% de los pacientes, y el 25% de los supervivientes presentan otras secuelas renales a largo plazo. La gravedad de la enfermedad aguda, particularmente del sistema nervioso central, y la necesidad de diálisis inicial están fuertemente asociadas con un peor pronóstico a largo plazo34. Puede haber casos familiares epidémicos, que coincidirán en el tiempo, a diferencia del SUHa. Los casos en adultos se asemejan al cuadro clínico de los niños, aunque suelen ser de mayor gravedad.
Síndrome urémico hemolítico atípicoClínicamente presentan datos similares al SUH típico, pero con mucha mayor gravedad a corto y largo plazo. El comienzo suele ser abrupto, aunque en un 20% de los pacientes puede ser progresivo (semanas o meses) con función renal conservada de inicio, anemia subclínica y trombocitopenia fluctuante35. Los pacientes con la forma familiar de la enfermedad tienen mal pronóstico, con una tasa de mortalidad del 10-15% y de IRC terminal de entre el 50 y el 80% tras el primer episodio agudo, pero varía según la anomalía subyacente20. Aunque el inicio se produce con la misma frecuencia en niños que en adultos, el pronóstico es diferente: en niños la mortalidad es superior, mientras que en los adultos es más frecuente la progresión a IRC36.
Entre el 60 y el 70% de los pacientes con mutaciones en el FHC, FIC o C3 y el 30% con autoanticuerpos anti-FHC fallecen durante el episodio inicial o desarrollan IRC. Las mutaciones en el FHC se asocian particularmente con pérdida de la función renal (88%). Los portadores de mutaciones en la PCM tienen mejor pronóstico, ya que aunque las recurrencias son frecuentes, se alcanza remisión completa en el 80 al 90% de los pacientes37. En aproximadamente un 10 a un 20% de los pacientes el trastorno se desarrolla durante el embarazo o puerperio36.
Puede haber afectación neurológica o multivisceral hasta en el 20% de los pacientes, aunque la primera es más global y menos fluctuante que en la PTT. Asimismo, son habituales los síntomas abdominales (dolor, náuseas, vómitos y diarrea) en la presentación aguda. Es frecuente la hipertensión arterial, de origen renal, así como las manifestaciones del aumento de la permeabilidad vascular, como derrame pleural, pericárdico, ascitis, anasarca y edema cerebral o pulmonar37. El SUHa puede reaparecer después del trasplante renal, y la tasa de recurrencia se asocia claramente con la carga genética. Los pacientes con mutaciones demostradas tienen 3 veces más posibilidades de recurrencia que los que no las tienen; asimismo, la presencia de mutaciones del FHC o de genes híbridos con FHC/FHCR1 y FBC/C3 se asocian 4 veces más a recurrencias que su ausencia38.
Diagnóstico diferencial de las microangiopatías trombóticasPor la gravedad del cuadro y por la importancia que tiene iniciar un pronto tratamiento, la primera entidad a considerar ante una MAT es la de una PTT adquirida (autoinmunitaria) o congénita. Sin embargo, el desarrollo de una MAT se puede relacionar con otras situaciones o enfermedades39, como bien ponen de manifiesto los datos del registro japonés de MAT, resumidos en la tabla 1 y donde se puede observar la distribución por frecuencia de las diferentes causas5. A lo largo de los últimos años se han ido describiendo diferentes mecanismos fisiopatológicos que determinan la aparición de una MAT en cada una de las diferentes situaciones, lo que debería permitir tener un enfoque terapéutico diferente40. Ello obliga a la realización de un diagnóstico diferencial entre las diferentes situaciones relacionadas con el desarrollo de una MAT (tabla 1).
En el diagnóstico diferencial de una MAT hoy en día sigue siendo esencial la historia clínica, que nos proporcionará valiosos datos sobre cómo enfocar el caso. Por ejemplo, los antecedentes de un tratamiento con ticlopidina o una neoplasia. Como se ha comentado previamente, son raros los pacientes que presentan la péntada clásica de la PTT.
La existencia de un antecedente de diarrea y afectación renal orienta al diagnóstico de un SUH secundario a una infección entérica producida por E. coli O157:H7, pero ante lo inespecífico del síntoma, se debe confirmar la sospecha mediante la detección de la toxina Shiga o los genes que la codifican en las heces o en el cultivo de estas41.
A continuación, las pruebas de laboratorio desempeñan un papel esencial (tabla 2). Sin embargo, ante un cuadro agudo, en general, el primer abanico de pruebas que tenemos a nuestra disposición es el que nos proporcionan los laboratorios de asistencia continuada. Los datos que proporcionan serán igualmente esenciales para orientar el caso. Y algo tan básico como una prueba de antiglobulina directa tiene una importancia enorme, pues puede cambiar la sospecha diagnóstica desde una MAT a un síndrome de Evans. En caso de que la MAT se acompañe de valores bajos de reticulocitos y un volumen corpuscular medio aumentado, quizá nos encontremos ante un déficit de vitamina B1242 o un defecto de cobalamina C, que cursa con valores aumentados de homocisteína, sin déficit de vitamina B1243.
En general, la trombocitopenia suele ser más profunda en los pacientes diagnosticados de PTT que en aquellos con otras causas de MAT. En una serie de 135 pacientes, los valores medios de la cifra de plaquetas en el momento del diagnóstico de PTT eran de 25,3±19,4×109/l (con unos límites entre 5 y 120×109/l)44. Los valores medios hallados en 65 pacientes con déficit grave de ADAMST13 (actividad<10%) fueron 16×109/l (con unos límites entre 2 y 124×109/l)45.
La presencia de esquistocitos, el sello característico de las MAT, no es exclusivo de ellas, pues se encuentran hasta en un 80% de los pacientes con una prótesis valvular o enfermedades renales, pero en cantidades inferiores a 0,5 por cada 100 hematíes. Sin embargo, la presencia de más de un un esquistocito por cada 100 hematíes en el frotis de sangre periférica es altamente indicativa de MAT46,47.
Pero, probablemente, la prueba que en los últimos años ha cambiado sustancialmente el diagnóstico diferencial inicial de una MAT es la posibilidad de disponer en un tiempo razonable del resultado de la actividad de ADAMTS13. Así, en ausencia de antecedentes de ingesta de ticlopidina o conectivopatías, una actividad inferior al 5-10% y la presencia de un inhibidor contra ADAMTS13 evidencia que nos encontramos ante un caso de PTT adquirida (autoinmunitaria)5.
En el caso de valores bajos de actividad de ADAMTS13 (<5-10%) pero sin inhibidor, las opciones diagnósticas pueden ser más amplias y, por ejemplo, tratarse de un déficit congénito de ADAMTS13. Conviene destacar que hay casos diagnosticados a los 45 años de edad32, pero también esporádicamente se pueden observar estos valores en las MAT asociadas a sepsis o a TPH48.
Unos valores no significativamente reducidos de ADAMTS13 (5-10%) aparecen fundamentalmente en las MAT secundarias y, más importante, en el SUHa. En general, las MAT secundarias se pueden excluir con las pruebas diagnósticas realizadas durante los primeros días tras el comienzo de la MAT. Sin embargo, en el caso del SUHa, las pruebas que nos confirmarán si nos encontramos ante una alteración de las moléculas reguladoras del complemento solo están al alcance de unos pocos laboratorios y pueden tardar semanas en llegar. Por esta razón, y por la existencia de un tratamiento específico para el SUHa, en la actualidad la actitud que se suele seguir es que si la actividad de ADAMTS13 es superior al 5-10%, existen signos de insuficiencia renal y se han descartado otras formas de MAT, se considera la MAT como SUHa mientras no se demuestre lo contrario40. En la figura 1 se resume una aproximación diagnóstica y terapéutica tras el diagnóstico de una MAT.

Recientemente ha sido publicado que los pacientes afectados de SUHa tienen unos mayores niveles de marcadores de activación del complemento terminal (C5a y C5b-9), significativamente superiores que los de pacientes con PTT49. Desgraciadamente el solapamiento de los valores observado entre los 2 grupos de pacientes limita seriamente la capacidad diagnóstica de dicha determinación.
Pruebas diagnósticas específicas de las microangiopatías trombóticasAnálisis de la metaloproteasa ADAMTS13Los laboratorios especializados pueden determinar 3 aspectos relacionados con el ADAMTS13: el antígeno, la actividad y la presencia de anticuerpos. Para llevar a cabo estas determinaciones es aconsejable utilizar muestras de plasma provenientes de muestras de sangre recogidas en citrato sódico como anticoagulante. Si no es posible procesar la muestra durante las primeras 4h tras la extracción, la muestra de plasma debe ser congelada a una temperatura≤−70°C, hasta un máximo de 6 meses, y debe descongelarse a 37°C en el momento de realizar las determinaciones.
Medida de la actividad ADAMTS13La actividad ADAMTS13 es el principal parámetro a evaluar por su valor informativo acerca de la funcionalidad de la molécula. Para ello, existen métodos indirectos que miden la actividad residual del FvW después de la acción de ADAMTS13, o directos que miden directamente la actividad de la metaloproteasa50. Los métodos aplicados inicialmente, o de primera generación, consisten en técnicas funcionales que se basan en la degradación de la molécula completa de FvW, plasmático o recombinante, por la enzima presente en la muestra de plasma a analizar. El grado de proteólisis puede ser medido por distintas técnicas, como el análisis multimérico del FvW (por resolución mediante SDS-agarosa o SDS-poliacrilamida y revelado tras transferencia proteica para visualizar las bandas)7,51, la unión del FvW al colágeno (collagen binding assay)52 o a la ristocetina (agregación inducida por ristocetina)53. Estos ensayos requieren someter al FvW a unas condiciones que permitan su desplegamiento, facilitando el acceso de la metaloproteasa al lugar de acción. Si el ADAMTS13 es funcional en la muestra en estudio, proteolizará el FvW utilizado como sustrato, resultando en una pérdida de multímeros de alto peso molecular y, como consecuencia, una menor unión al colágeno y una disminución en la agregación inducida por ristocetina.
Los métodos más recientes, o de última generación, emplean fragmentos de la molécula de FvW. La utilización del dominio VWFA254 o del fragmento VWF7355,56 del FvW, que contienen el lugar de acción para ADAMTS13, permite aumentar la especificidad, la sensibilidad y la reproducibilidad de la técnica, pues al añadir estos fragmentos se elimina la variabilidad debida a la composición multimérica y a la cantidad del FvW añadida como sustrato. Al mismo tiempo no requieren un tratamiento previo para facilitar la acción enzimática de ADAMTS13. Existen técnicas de enzimoinmunoanálisis que utilizan estos fragmentos, conjugados con marcadores, inmovilizados en placas sobre las que se añade la muestra de plasma en la que se quiere determinar la actividad de ADAMTS13. La cantidad de sustrato residual será inversamente proporcional a la actividad ADAMTS13 de la muestra.
La incorporación de un marcador fluorescente a estos fragmentos y su detección basada en técnicas de transmisión de energía fluorescencia/resonancia permite obtener resultados en aproximadamente una hora. El fragmento VWF73, derivado del dominio A2 del FvW, ha sido modificado químicamente de manera que contiene un fluoróforo y una molécula que actúa inhibiendo este57. Cuando ADAMTS13 actúa sobre este fragmento, separa ambas moléculas, de manera que el fluoróforo emite fluorescencia cuando es expuesto a una cierta longitud de onda. La fluorescencia emitida, que es registrada durante un tiempo determinado, es directamente proporcional a la actividad de ADAMTS13.
Por otro lado, puede ser interesante determinar la cantidad de antígeno en aquellos casos en los que la actividad es nula y no se detecten anticuerpos anti-ADAMTS13, para comprobar si las alteraciones congénitas afectan la expresión de la proteína o su actividad funcional. La mayoría de las técnicas descritas para la determinación de la actividad y la cantidad de antígeno están actualmente disponibles comercialmente. Los valores de normalidad de la actividad ADAMTS13 son muy variables y se han definido intervalos de referencia que pueden variar dependiendo de la técnica utilizada y del laboratorio. Los valores de actividad ADAMTS13 en individuos sanos están entre 90 y 110%, pero existen estudios que sitúan la normalidad por encima del 50%58. Así mismo, la experiencia acumulada tras analizar un número considerable de muestras del registro español permite afirmar que en aquellos pacientes en los que se ha llegado al diagnóstico de PTT, los valores de actividad ADAMTS13 son<5%24, aunque otros laboratorios de referencia establecen como valor de corte el 10% de la actividad58.
Es interesante destacar que la medida de actividad de ADAMTS13 y de la presencia de anticuerpos anti-ADAMTS13 es útil para predecir la recurrencia de la PTT. Los supervivientes de un episodio agudo de PTT adquirida con niveles de actividad muy reducidos y/o presencia de anticuerpos anti-ADAMTS13 durante la remisión tienen aproximadamente 3 veces mayor probabilidad de desarrollar un nuevo episodio de PTT que los pacientes con una mayor actividad de la proteasa y sin anticuerpos59.
Determinación de anticuerpos inhibidores de la actividad ADAMTS13El 90% (o más) de los casos de PTT adquirida, con actividad ADAMTS13<5%, están asociados a la presencia de anticuerpos anti-ADAMTS1351,60. Los anticuerpos mayoritarios son IgG, principalmente subclase IgG4, seguida por IgG1, IgG2 e IgG3. La presencia de autoanticuerpos con acción inhibitoria puede ser detectada mediante la técnica utilizada para medir actividad ADAMTS13 en una muestra en la que se mezcla plasma del paciente con plasma control (1:1). La capacidad inhibitoria del anticuerpo en la muestra de plasma se mide en unidades Bethesda (una unidad de inhibidor corresponde a la concentración de anticuerpo capaz de inhibir la actividad ADAMTS13 en un 50%). Pero, además, existen anticuerpos que no son neutralizantes, pero que afectan la eliminación de ADAMTS13 de la circulación. Actualmente, existen pruebas de enzimoinmunoanálisis comerciales que utilizan ADAMTS13 recombinante y facilitan la detección de anticuerpos anti-ADAMTS1361. Estas técnicas proporcionan resultados expresados como U/ml, calculadas con base en una recta patrón. Debe tenerse en cuenta que también se han descrito IgM e IgA anti-ADAMTS13.
Otras pruebas diagnósticasDetección de infección por Escherichia coli productora de toxina similar a Shiga o verotoxinaExisten cepas de E. coli capaces de producir toxinas similares a las que produce S. dysenteriae, denominadas toxinas Shiga. Se han descrito 2 toxinas (similares a Shiga o verotoxinas): toxina Shiga 1 (stx1) y toxina Shiga 2 (stx2). Estas toxinas son responsables del desarrollo de SUH62. Para su detección, tradicionalmente se aplican técnicas de cultivo microbiológico (en medio McConkey con sorbitol), reacción en cadena de la polimerasa utilizando oligonucleótidos específicos que amplifican fragmentos de los genes de stx1 y stx263, pruebas serológicas para la detección de anticuerpos contra estas toxinas y la medida de enterohemolisina64. Existe actualmente una prueba comercial rápida (Shiga Toxin Quick Chek test, Alere™) capaz de detectar toxinas E. coli productoras de verotoxina/STEC directamente en muestras fecales, incluso de aquellas cepas de E. coli que no son detectadas por los métodos tradicionales.
Determinación de proteínas del sistema complementoMutaciones en uno o más genes que codifican proteínas reguladoras (FHC, FIC, PCM, trombomodulina) y proteínas activadoras (factores B y C3) del sistema del complemento pueden dar lugar a SUHa. También se han detectado casos de SUHa asociados al desarrollo de anticuerpos anti-factor H. La medida de valores séricos de C3, C4, FHC y FIC, así como de la expresión de PCM en células mononucleares de sangre periférica (citometría de flujo), de anticuerpos anti-factor H, y el cribado de posibles mutaciones que afecten a los genes mencionados podría ayudar a determinar el diagnóstico directo de esta enfermedad22,65.
Tratamiento de la púrpura trombocitopénica trombótica adquirida y congénitaPúrpura trombocitopénica trombótica adquiridaTratamiento de primera líneaRecambio plasmáticoEl recambio plasmático (RP) permite la eliminación de los anticuerpos dirigidos contra ADAMTS13 y, mediante la solución de reposición, aportar la metaloproteasa deficitaria. Actualmente existe evidencia científica suficiente como para afirmar que el tratamiento de primera línea de la PTT adquirida idiopática es el RP66,67, aunque algunos aspectos relacionados con el esquema terapéutico aún no están totalmente establecidos.
Momento de inicio del RP. La PTT debe ser considerada una urgencia hematológica y el RP debe iniciarse lo antes posible. Ante una anemia hemolítica microangiopática y trombocitopenia, en ausencia de otras causas, debe sospecharse una PTT e iniciar el RP inmediatamente, preferiblemente antes de transcurridas 4-8h desde el diagnóstico68, ya que el 50% de las muertes por PTT se producen durante las primeras 24h, y esta mortalidad con frecuencia está relacionada con un retraso en el inicio del RP. Solo en el caso de que no se pudiera realizar el RP se iniciaría la infusión de plasma a dosis de 25-30ml/kg69.
Volumen a recambiar. Por debajo de un volumen plasmático la eficacia del RP es escasa y por encima de 2 volúmenes plasmáticos los efectos adversos se incrementan. Un esquema terapéutico que se utiliza con frecuencia es iniciar el recambio de 1,5 volemias plasmáticas/día hasta lograr una estabilización en los parámetros clínicos, y posteriormente disminuir a una volemia plasmática. En el caso de riesgo vital se puede incrementar a 2 volemias plasmáticas o realizar un RP cada 12h, aunque esta práctica se utiliza cada vez menos, ya que suele ser más habitual introducir precozmente otros fármacos inmunodepresores45,70.
Solución de reposición. Se han usado diversos tipos de plasma como solución de reposición: plasma fresco congelado (PFC) no modificado, sobrenadante de crioprecipitado o plasma inactivado por diversos métodos (solvente detergente, azul de metileno, amotosaleno). El nivel de actividad de ADAMTS13 es similar en todas las soluciones de reposición, aunque ligeramente superior en el plasma no modificado y en el sobrenadante de crioprecipitado. La actividad de ADAMTS13 no disminuye en productos plasmáticos descongelados y mantenidos entre 1-6°C hasta 5 días71. Aunque en un momento se planteó la superioridad del uso de sobrenadante de crioprecipitado, por carecer de multímeros de FvW de muy alto peso molecular, actualmente se sabe que no es así72,73.
A la vista de los conocimientos actuales, no se puede afirmar la superioridad de una solución de reposición en relación con las otras, excepto en el caso del plasma inactivado con azul de metileno, ya que diferentes estudios publicados lo han relacionado con un mayor número de RP para alcanzar la remisión y con un aumento del consumo de plasma74. Se han descrito reacciones anafilácticas IgE específicas asociadas al plasma inactivado con azul de metileno. Sin embargo, cuando se compara la incidencia de reacciones alérgicas comunicadas a los sistemas de hemovigilancia observadas con el plasma tratado con azul de metileno no son diferentes a las recogidas con otros tipos de plasma75. Algunos autores apuntan la posibilidad de usar albúmina al 5% durante la primera mitad del procedimiento, y plasma en el resto76.
Número de sesiones. Se debe continuar realizando RP diariamente hasta 2 días después de alcanzar la remisión completa, entendiendo como tal un recuento de plaquetas superior a 150×109/l. Hay que tener en cuenta que en la población española el recuento de plaquetas suele ser inferior, por lo que se podría reducir el límite a 125×109/l77. Una práctica frecuente es la reducción progresiva de la frecuencia de los recambios o del volumen a recambiar una vez alcanzada la remisión completa, pero no se ha demostrado hasta ahora que ninguna de las 2 influya en la prevención de recaídas78.
CorticoidesEl fundamento de su uso en el tratamiento de la PTT adquirida reside en la supresión de la producción de anticuerpos anti-ADAMTS1348. Aunque su uso se ha generalizado, el único estudio clínico que sustenta la utilización de corticoides sería el publicado por Bell et al. en 1991. Los autores describieron la experiencia del John Hopkins Hospital en el tratamiento de pacientes con PTT-SUH. Treinta enfermos recibieron prednisona y 78 prednisona combinada con RP. Aunque los resultados son mejores en el grupo de RP más corticoides, más de la mitad de los pacientes que reciben únicamente tratamiento con corticoides alcanzan la remisión, aunque dichos pacientes mostraban formas leves de la enfermedad79.
La dosis de prednisolona combinada con los RP generalmente recomendada es de 1mg/kg/día68, aunque un estudio aleatorizado comparativo encontró que dosis mayores (metilprednisolona 10mg/kg/día durante 3 días y posteriormente 2,5mg/kg/día) a los 23 días de tratamiento consiguen mayores tasas de respuesta (46,6 frente a 32%)80. El incremento del uso de los corticoides en el tratamiento de las MAT se asocia con una reducción del número de RP necesarios para alcanzar la remisión y, como consecuencia de este hecho, con una menor morbilidad asociada a los RP48.
Otros tratamientosRituximabRituximab es un anticuerpo monoclonal anti-CD20 que determina una eliminación de linfocitos B en la sangre periférica. Aunque su indicación principal es en el tratamiento de linfomas no hodgkinianos y otros procesos linfoproliferativos de estirpe B, ha demostrado su eficacia en el tratamiento de enfermedades con base autoinmunitaria, como: artritis reumatoide, púrpura trombocitopénica inmunitaria, anemia hemolítica autoinmunitaria y hemofilia adquirida81. Por este motivo, se ha propuesto su utilización en el tratamiento de la PTT.
Rituximab en PTT refractaria o en recaída. El uso de rituximab se ha demostrado eficaz en el tratamiento de PTT refractarias82 o recidivas precoces83. El uso de rituximab durante el tratamiento de la PTT se ha relacionado con un menor riesgo de recaídas84. Se ha propuesto el uso de rituximab en pacientes con antecedentes de PTT, en una situación clínica de recaída pero con valores de actividad de ADAMTS13<6% y un título alto de anticuerpos inhibidores85. Sin embargo, no está demostrado que la existencia de unos valores bajos de actividad de ADAMTS13 se relacionen con recaídas86, por lo que la determinación analítica de la actividad no debe ser usada como indicación de tratamiento en ausencia de manifestaciones clínicas o biológicas45.
Rituximab en primera línea. Recientemente se han publicado trabajos que valoran el empleo de rituximab en etapas precoces de la enfermedad82,87. Los datos apuntan a que el uso de rituximab se asocia con una menor tasa de recidivas y una más pronta respuesta, sin que exista un incremento de las infecciones. En las guías clínicas británicas se recomienda su uso precoz, junto con RP y corticoides, en aquellos pacientes con alto riesgo de mortalidad (afectación neurológica y/o cardiaca)68.
Cuestiones prácticas. Rituximab debe administrarse al finalizar el RP. La realización de RP el día después no afecta la efectividad de rituximab88. No existe una recomendación específica de dosis para los pacientes con PTT, aunque suele utilizars la misma que en los síndromes linfoproliferativos, 375mg/m2/semana durante 4 semanas. Teniendo en cuenta el carácter no proliferativo de la PTT, algunos grupos preconizan el empleo de dosis más bajas, al igual que se utiliza en otras enfermedades autoinmunitarias, aunque no existen ensayos clínicos que permitan establecer una recomendación al respecto89,90.
AntiagregantesEn la actualidad no hay evidencia científica que avale su uso rutinario. Se ha descrito un incremento de hemorragias asociadas a su uso, por lo que solo deben administrarse si el recuento de plaquetas es>50×109/l68.
EsplenectomíaSe ha propuesto con objeto de disminuir las recidivas, aunque no está clara su eficacia68.
Otros inmunodepresoresSe han utilizado diversos agentes inmunodepresores, como la vincristina, la azatioprina o la ciclofosfamida, en pacientes refractarios, con resultados satisfactorios en algunos casos91. Aunque el uso de ciclosporina se ha relacionado con el desarrollo de MAT, se ha demostrado su eficacia en casos refractarios al tratamiento con RP con recuperación de los valores de actividad de ADAMTS13 y desaparición de inhibidores92. También ha mostrado una cierta eficacia previniendo las recaídas93. La dosis propuesta es de 2-3mg/kg/día en 2 dosis por vía oral.
Otras estrategias terapéuticasADAMTS13 recombinante. El fundamento sería restaurar la actividad ADAMTS13 y actualmente está en fase de uso experimental. Ensayos en laboratorio y en el animal de experimentación han demostrado la eficacia del uso de ADAMTS13 recombinante en PTT congénitas y adquiridas94,95. Los últimos trabajos publicados evidencian que el fabricante de la molécula actualmente está dirigiendo su desarrollo más hacia el potencial efecto protector del endotelio de ADAMTS13 que hacia el tratamiento de la PTT96.
N-acetilcisteína. El uso de N-acetilcisteína se ha propuesto debido a su acción reductora de los puentes disulfuros de los multímetros del FvW97,98.
Anticuerpo anti-FvW. Actualmente está en marcha un ensayo clínico fase ii (TITAN) sobre el empleo de anticuerpos anti-FvW (NTC 01151423).
Inhibición de la unión de las plaquetas al FvW. En fases preliminares de investigación están el aptámero ARC1779 y el nanoanticuerpo ALX068199.
Tratamiento de soporteTransfusión de plaquetas. En los pacientes con PTT la trombocitopenia grave no es indicación de transfusión, pero debe considerarse en situación hemorrágica o para prevenir la pérdida de sangre antes de maniobras diagnósticas o terapéuticas con riesgo hemorrágico100.
Púrpura trombocitopénica trombótica congénitaEstá indicada la infusión de plasma. En estos casos, se puede administrar plasma a una dosis de 10-15ml/kg o concentrados de factor viii de pureza intermedia, donde se ha demostrado la existencia de actividad de ADAMTS13. La frecuencia y dosificación varía según el fenotipo del paciente68.
Tratamiento del síndrome urémico hemolítico atípico y otras microangiopatías trombóticasTras el diagnóstico de un SUHa, durante mucho tiempo la única opción terapéutica a considerar era el tratamiento con PFC, en forma de RP, o de infusión si este no era posible. Sin embargo, a medida que ha aumentado nuestro conocimiento de la fisiopatología del cuadro, el papel del RP en el tratamiento ha ido cambiando. Este hecho queda perfectamente reflejado en las distintas ediciones de la guía de uso de la aféresis terapéutica de la American Society for Apheresis (ASFA, «Asociación Americana de Aféresis»)101–103. En la sexta edición, el SUHa es dividido en 3 entidades con muy diferentes grados de indicación. En los casos donde se encuentren mutaciones de los genes del complemento se recomienda el RP como categoría ii (tratamiento de segunda línea, aislado o asociado a otros tratamientos). Sin embargo, cuando la mutación afecta a la PCM se considera categoría iv (definida como que se considera la aféresis inefectiva o perjudicial) debido a que esta proteína no se encuentra en plasma. Por el contrario, las formas debidas a la existencia de anticuerpos contra el FHC se consideran como una categoría i (definida como que la aféresis es el tratamiento de primera línea, aislada o asociada a otras terapéuticas), combinada con inmunodepresores como corticoides o rituximab104.
A pesar de utilizar el RP en el tratamiento del SUHa, la tasa de respuesta es muy variable dependiendo de la causa subyacente. Por ejemplo, en el caso de defectos que afecten al FHC, el 43% de los pacientes tratados con RP fallecen o entran en IRC65,105. Además, el tratamiento se ve frecuentemente complicado por la aparición de reacciones alérgicas al PFC, y en los pacientes pediátricos presenta desafíos importantes en cuanto al abordaje de accesos venosos para el RP y la realización del proceso.
Afortunadamente, a la vez que se identificaba la activación inapropiada del complemento como la causa subyacente de la aparición del SUHa, se estaba desarrollando un anticuerpo monoclonal para bloquear dicha activación in vivo, aunque con un objetivo completamente diferente. En 1995 se identificó un anticuerpo monoclonal murino capaz de inhibir la conversión de C5 a C5a. Posteriormente dicho anticuerpo se modificó mediante bioingeniería para transformarlo en un anticuerpo quimérico humanizado para reducir su inmunogenicidad y las funciones efectoras106. Dicho anticuerpo, eculizumab (Soliris®, Alexion), fue aprobado en 2007 en Europa y EE. UU. para el tratamiento de los pacientes con hemoglobinuria nocturna paroxística. Sin embargo, su mecanismo de acción pronto despertó interés por su uso entre los profesionales que trataban el SUHa. En efecto, se empezaron a publicar casos de pacientes con SUH refractarios al tratamiento convencional de RP que respondían a la administración de eculizumab en formas primarias107 o con recaídas tras trasplante de riñón108. Eculizumab fue autorizado para el tratamiento del SUHa tanto en pacientes pediátricos como en adultos en primera línea por la Administración de Alimentos y Fármacos estadounidense y por la Agencia Europea de Medicamentos en septiembre y noviembre de 2011, respectivamente.
Recientemente se han publicado 2 estudios prospectivos de la administración de eculizumab en pacientes con SUHa. La administración de eculizumab permitió suspender el tratamiento con PFC en el 88 y en el 100% de los pacientes del estudio 1 y 2, respectivamente, con la desaparición de signos de actividad de la enfermedad (trombocitopenia o signos de anemia microangiopática). Además, 4 de los 5 pacientes que estaban en diálisis recuperaron la función renal, lo que permitió interrumpir dicho procedimiento. El tratamiento con eculizumab de 62 a 64 semanas no se asoció a episodios adversos mayores109. Hay que destacar que previamente al inicio del tratamiento con eculizumab el paciente debe ser vacunado frente a Neisseria meningitidis.
En el caso de pacientes pediátricos, y dado que esta vacuna no protege contra el serotipo B, que, además, es el más prevalente, se recomienda la administración de profilaxis antibiótica con penicilina o amoxicilina mientras dure el tratamiento con eculizumab, así como vacunación frente a Haemophilus influenzae y neumococo110.
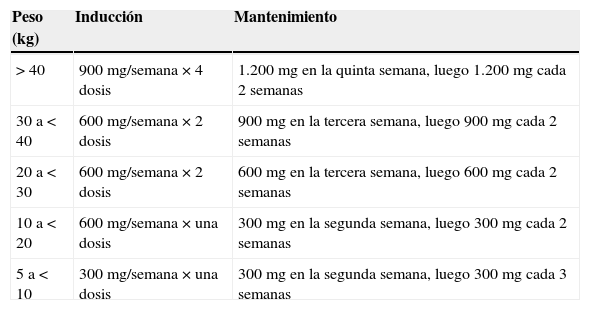
El precio elevado de eculizumab (un año de tratamiento con 1.200mg cada 2 semanas asciende a unos 290.000€ según la última actualización de precio aprobada por el Ministerio en septiembre de 2013) está limitando su uso generalizado. Es por ello que en la actualidad la mayoría de las guías recomienda que, en los pacientes pediátricos, una vez descartado el SUH asociado a toxina Shiga, se debe iniciar eculizumab tan pronto como sea posible, según se indica en la tabla 4. Un óptimo bloqueo del complemento es esencial en la fase aguda del SUHa (ver tabla 4 para dosificación). La prueba del CH50 puede ser útil para monitorizar el nivel de bloqueo del complemento111, aunque no existe unanimidad sobre su utilización.
Dosificación de eculizumab en pacientes menores de 18 años con síndrome urémico hemolítico atípico
| Peso (kg) | Inducción | Mantenimiento |
|---|---|---|
| >40 | 900mg/semana×4 dosis | 1.200mg en la quinta semana, luego 1.200mg cada 2 semanas |
| 30 a<40 | 600mg/semana×2 dosis | 900mg en la tercera semana, luego 900mg cada 2 semanas |
| 20 a<30 | 600mg/semana×2 dosis | 600mg en la tercera semana, luego 600mg cada 2 semanas |
| 10 a<20 | 600mg/semana×una dosis | 300mg en la segunda semana, luego 300mg cada 2 semanas |
| 5 a<10 | 300mg/semana×una dosis | 300mg en la segunda semana, luego 300mg cada 3 semanas |
Fuente: Keating110.
Sin embargo, a diferencia de los niños, el diagnóstico de SUHa en un adulto que presenta un primer episodio de MAT es un reto mucho mayor y requiere un número superior de pruebas para alcanzar el diagnóstico. En general se recomienda iniciar RP con PFC como solución de reposición mientras se completa el diagnóstico. Si tras el tratamiento inicial se han descartado otras formas de MAT y no hay respuesta (definida como persistencia de la trombocitopenia y/o persistencia de LDH elevada y/o no disminución de la creatinina sérica en más de un 25%) se debe iniciar eculizumab (fig. 1)111. Conviene destacar que se ha descrito un polimorfismo en un único nucleótido de C5 que lo hace resistente al bloqueo con eculizumab y que se ha encontrado en un 3,5% de la población nipona112.
Un aspecto pendiente de definir con claridad es si se debe interrumpir el tratamiento con eculizumab en algún momento de la evolución de la enfermedad. Algunos autores preconizan no interrumpirlo antes del año desde el primer episodio para minimizar el riesgo de recaídas111. Si se interrumpe el tratamiento, se recomienda extremar los controles para la detección precoz de una posible aparición de un nuevo episodio de SUHa113.
Es de destacar cómo recientemente se ha publicado el uso de eculizumab en casos de brotes de SUH asociados a STEC. Delmas et al. han utilizado eculizaumb en 9 pacientes que formaban parte de un brote epidémico, entre 0 y 4 días después del diagnóstico; 2 de ellos requerían diálisis. Los pacientes recibieron 900mg por semana durante 4 semanas y posteriormente 1.200mg las semanas 5, 7 y 9. Hubo un rápido control de los signos de MAT con normalización en 7 días del recuento plaquetario en todos los pacientes, así como una normalización del filtrado glomerular estimado a las 10 semanas también en todos ellos41. Estos resultados entran en conflicto con los descritos por autores alemanes durante el mismo brote114,115. Probablemente, el momento de iniciar el tratamiento con eculizumab (precoz o tardío) y la realización hasta en un 40% de los pacientes de RP tras la administración del fármaco en el grupo alemán puedan explicar la diferente respuesta al tratamiento.
Respecto a la MAT asociada al TPH, la terapéutica actual es empírica y ninguna de las diferentes opciones empleadas se ha demostrado especialmente útil116. La eficacia de los RP en este cuadro es incierta y no está exenta de complicaciones; de hecho, su indicación es una categoría iii en la guía de la ASFA (definida como que el tratamiento con aféresis no está establecido y la indicación de realizarla debe individualizarse)103. Dada la íntima asociación entre la aparición de MAT y el uso de inhibidores de la calcineurina, la mayoría de los autores coinciden en que su sustitución por otros fármacos (micofenolato de mofetilo, esteroides) forma parte esencial del control de esta complicación. Otros tratamientos empleados ocasionalmente con resultados dispares en estos pacientes son rituximab, daclizumab, defibrotido y ácido eicosapentaenoico117–119. Recientemente se ha reportado el hallazgo de una alta prevalencia de mutaciones en los genes que codifican para el CFH en niños que presentan una MAT pos-TPH, que evidencia que una activación inadecuada del complemento podría estar involucrada en la fisiopatología del cuadro120. El mismo grupo publicó posteriormente que el tratamiento con eculizumab, a dosis superiores de las habitualmente requeridas en el tratamiento del SUHa, fue efectivo en 4 de 6 pacientes pediátricos, eliminando las manifestaciones de MAT121. El tratamiento de un adulto con una MAT pos-TPH con eculizumab indujo igualmente la respuesta122.
El tratamiento de otras formas de MAT se basa fundamentalmente en tratar la causa subyacente. Así, puede requerir desde tratar la infección por el virus de la inmunodeficiencia humana123, hasta tratar el cáncer metastásico subyacente o incluso administrar vitamina B12 para tratar la anemia perniciosa excepcionalmente asociada a MAT124.
ConclusionesEl diagnóstico y tratamiento de las MAT ha cambiado significativamente en los últimos años. Nuestro mejor conocimiento de la fisiopatología ha hecho que se haya pasado de un diagnóstico sindrómico y un tratamiento similar en todos los casos a buscar un diagnóstico etiológico y un tratamiento específico de la entidad subyacente provocadora del síndrome.
Evidentemente ello ha aumentado la complejidad del proceso diagnóstico y hoy en día algunos casos representan un auténtico desafío para los profesionales a cargo del paciente. Sin embargo, afortunadamente, no solo tenemos a nuestra disposición un amplio abanico de pruebas de laboratorio que nos dan un apoyo esencial a la hora del diagnóstico, sino que también en los últimos años hemos ampliado la panoplia terapéutica para tratarlas.
El resultado de todo ello es que actualmente somos capaces de ofrecer a nuestros pacientes un mejor pronóstico de su enfermedad con una menor iatrogenia. Desafortunadamente en algunos casos el coste del tratamiento representa una barrera difícil de sortear y realmente esperamos y deseamos que en el futuro ese obstáculo se pueda reducir significativamente.
Sin embargo, aún existe un pequeño porcentaje de pacientes con MAT en los cuales el pronóstico es malo, e incluso infausto. Ello nos debe servir de recordatorio de que aún no hemos sido capaces de desentrañar todos los mecanismos implicados en la aparición de la enfermedad y diseñar el mejor tratamiento. La búsqueda de un mejor diagnóstico y unos tratamientos aún más eficaces debe continuar, y los autores estaríamos más que satisfechos si se pudiera considerar esta guía como un paso más en ese camino.
Conflicto de interesesEl Dr. Contreras ha impartido sesiones patrocinadas por Alexion Pharmaceuticals.
El Dr. de la Rubia ha impartido sesiones patrocinadas y ha realizado consultoría para Alexion Pharmaceutical.
El Dr. Del Río-Garma ha recibido una beca de investigación de Roche para el GEA.
La Dra. Díaz-Ricart declara no tener conflicto de intereses.
El Dr. García Gala ha impartido sesiones patrocinadas y ha realizado consultoría para Johnson&Johnson, Sandoz, y Alexión Pharmaceuticals.
El Dr. Lozano ha recibido becas de investigación de TerumoBCT, ha impartido sesiones patrocinadas por TerumoBCT y Alexion Pharmaceuticals, y ha realizado consultoría para Alexion Pharmaceuticals.

