




Introducción
Los ensayos clínicos aleatorizados (ECA) bien diseñados y correctamente ejecutados proporcionan la mejor evidencia sobre el efecto de las intervenciones sanitarias, pero una metodología inadecuada puede resultar en estimaciones exageradas de este efecto1,2.
Para poder evaluar la metodología de un ECA a partir de su publicación, es preciso que su diseño, ejecución y análisis se describan detalladamente, con exactitud y con transparencia. Sin embargo, se ha constatado repetidamente que la información facilitada en las publicaciones es muchas veces insuficiente3-6, o inexacta (por abuso de términos como análisis por intención de tratar7, o aleatorización8), y que ciertos problemas metodológicos son frecuentes9-11.
Con ánimo de mejorar la situación, a principios de los años noventa, dos grupos de editores, profesionales del ensayo clínico y metodólogos, elaboraron separadamente recomendaciones para comunicar ensayos clínicos12,13. Posteriormente, ambos grupos reunieron sus esfuerzos y consensuaron una propuesta conjunta que se publicó con el título de CONSORT statement, acrónimo de Consolidated Standards of Reporting Trials14. CONSORT es básicamente una lista-guía de los puntos que se consideran críticos y que por tanto deberían incluirse en todo informe o comunicación de un ensayo clínico, y un diagrama para ilustrar el flujo de individuos participantes durante el ensayo. En principio, la guía CONSORT se desarrolló para ensayos clínicos de grupos paralelos y, aunque gran parte de su contenido es también válido para otros diseños, se trabaja en extensiones o adaptaciones para otro tipo de ensayos15,16.
El objetivo de la guía CONSORT es facilitar la lectura crítica y la interpretación de los ECA, orientando a los autores, revisores y editores acerca de cómo debe comunicarse este tipo de estudios. Desde su publicación en 1996, diversas revistas y grupos editoriales (incluido el grupo de Vancouver) han adoptado la guía CONSORT, y posteriormente se ha verificado un aumento en la calidad de la comunicación de los ECA17,18. Sin embargo, también se puso de manifiesto ciertas limitaciones19, por lo que en 1999 se emprendió la revisión de la versión inicial, lo que dio lugar a una versión revisada que se publicó en 200120-22.
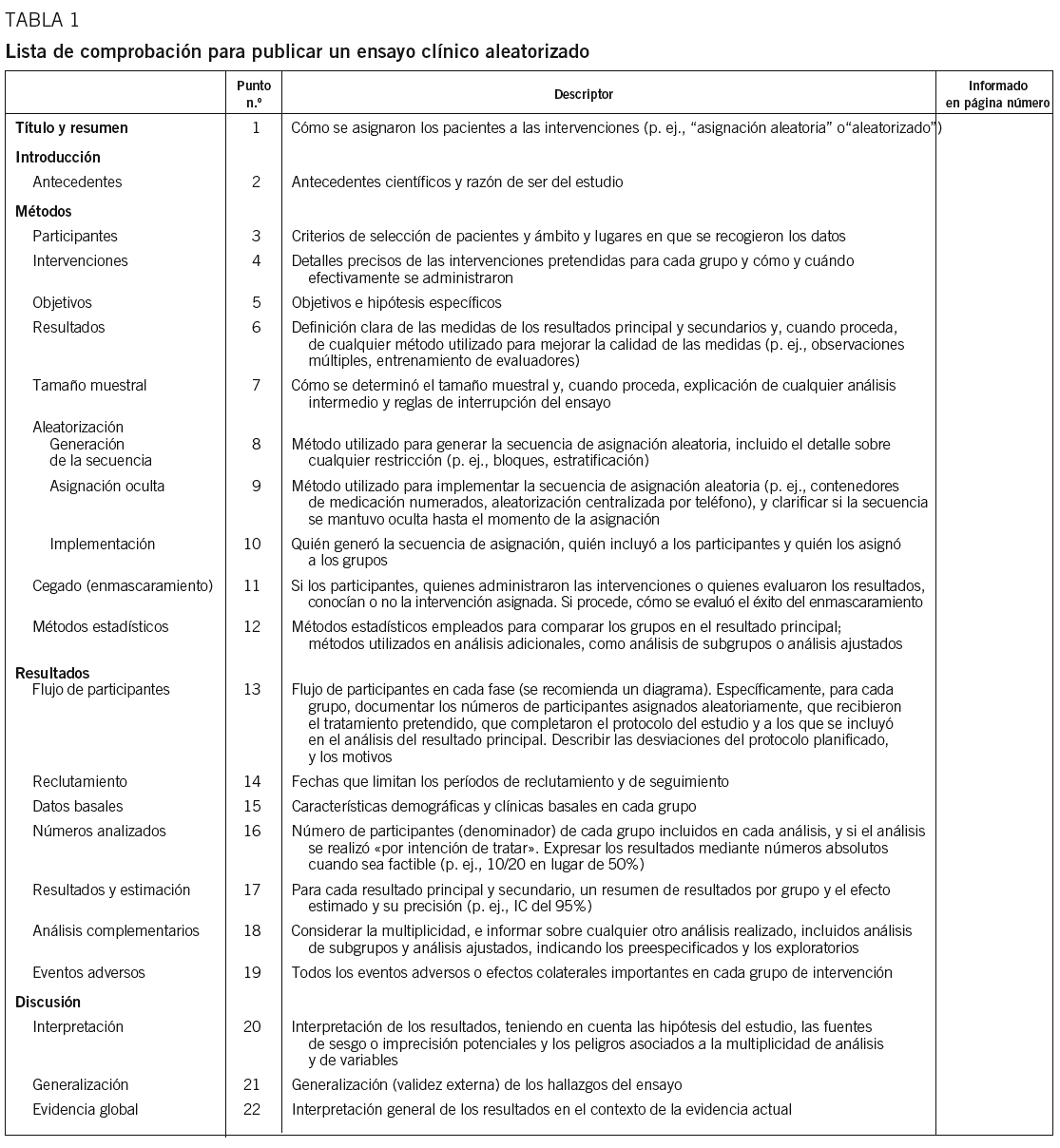
En este artículo se presenta la traducción de los 22 puntos (tabla 1) y del diagrama de flujo (fig. 1) que constituyen la guía CONSORT, acompañados de un breve comentario. En una publicación previa23 y también en internet (http:// www.consort-statement.org) se puede consultar la justificación de los 22 puntos incluidos en la guía, así como ejemplos publicados de lo que se consideran comunicaciones correctas.


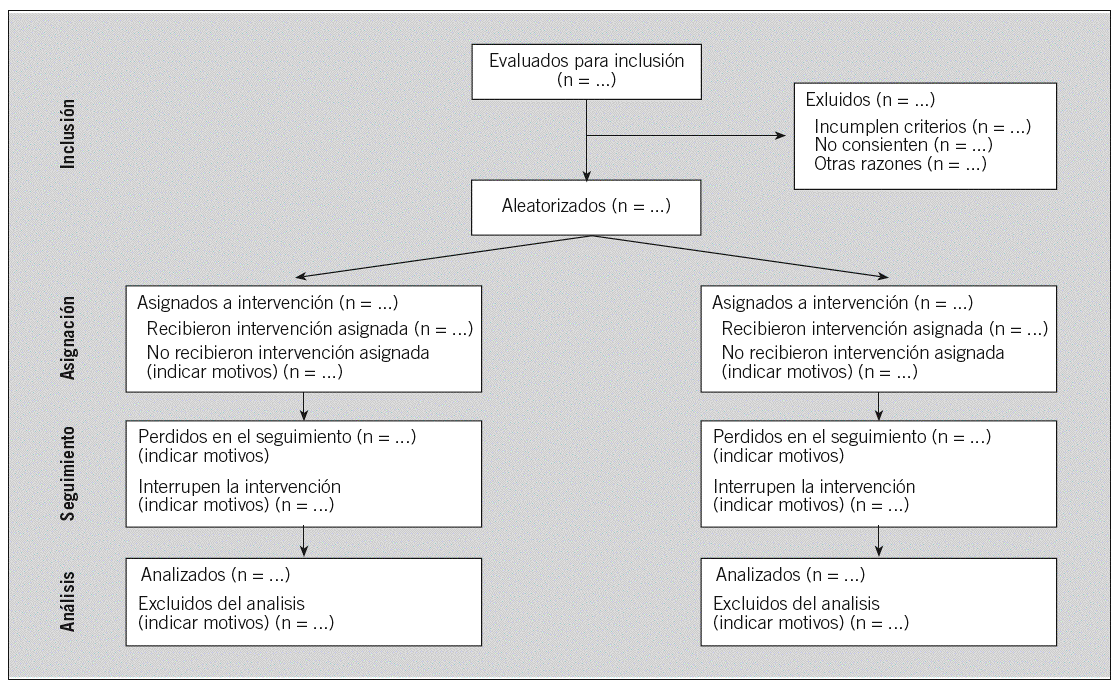
Fig. 1. Diagrama de flujo de participantes.
Puntos de la lista de comprobación
Título y resumen
1. Título y resumen. Para facilitar su indexación como ECA en las bases de datos bibliográficas y su identificación en las búsquedas24, se recomienda mencionar el método de asignación de los individuos a los grupos de tratamiento tanto en el resumen como en el título del artículo.
Introducción
2. Antecedentes. Resumir y referenciar el conocimiento previo, así como razonar la necesidad del ensayo, son condiciones para cumplir con el principio ético de evitar la exposición innecesaria de los individuos a los riesgos de una investigación poco justificada25. Algunos ensayos clínicos podrían haberse evitado haciendo una revisión sistemática de los previos26. Por ello, la introducción debe hacer referencia a los ensayos clínicos previos similares y a posibles revisiones sistemáticas de dichos ensayos, o constatar su ausencia.
Métodos
3. Participantes. En los ensayos clínicos, la población de individuos definida por la condición clínica de interés suele restringirse mediante criterios de selección tales como la edad, la ausencia de comorbilidad o de comedicación. Todos los criterios de selección (la distinción entre criterios de inclusión y de exclusión es innecesaria27) utilizados deben definirse explícitamente, así como los métodos utilizados para reclutar a los pacientes. Además de los criterios de selección, debe especificarse el ámbito y los centros en que se ha realizado el ensayo, porque pueden ser determinantes de la validez externa (posibilidad de generalización) de los resultados.
4. Intervenciones. Debe describirse las características de todas las intervenciones, incluidas las utilizadas como control (p. ej., placebo o práctica clínica habitual), particularmente las características de las que, presumiblemente, pueda depender el efecto. Dada la diversidad de posibles intervenciones (fármacos, cirugía, programas educativos, etc.), es difícil estandarizar la descripción de las características relevantes. En algunos casos puede ser imprescindible describir el entrenamiento o la experiencia de quienes practicaron la intervención, además de la técnica empleada (p. ej., intervenciones quirúrgicas)28. Si se trata de intervenciones complejas, como combinaciones de tratamientos, hay que describir los distintos componentes.
5. Objetivos. Debe mencionarse explícitamente los objetivos (o las correspondientes hipótesis) específicos del ensayo y, en el caso de los ensayos confirmatorios, es necesario distinguir el principal de los secundarios.
6. Resultados. El término resultados se refiere aquí a las variables utilizadas para evaluar comparativamente las intervenciones. Es necesario definir las variables utilizadas y distinguir la principal de las secundarias. La variable principal debe ser la que se consideró más importante al diseñar el ensayo y se utilizó para determinar el tamaño muestral. Es fundamental que su definición sea inequívoca: cuando la misma variable se evalúa repetidamente (distintos tiempos o métodos), hay que aclarar cuál de ellas, o qué combinación de ellas, es la que se consideró principal. En general, es recomendable definir una única variable principal para evitar problemas de multiplicidad (véase el punto 18); pero si hay más de una, es necesario tenerlos en cuenta y discutirlos. Si los hay, debería emplearse instrumentos (incluyendo escalas29, p. ej. de calidad de vida) y procedimientos de medida descritos previamente30, e idealmente consensuados, estandarizados o, al menos, de propiedades conocidas, describiendo estas propiedades31.
7. Tamaño muestral. Cuando se diseña un ensayo clínico, es ética y científicamente importante asegurar que el análisis principal tendrá una potencia adecuada, es decir, una probabilidad razonablemente alta de demostrar diferencias clínicamente relevantes si éstas existen. Por ello, es importante describir cómo se determinó el tamaño muestral, incluidos las estimaciones utilizadas en los cálculos y los posibles ajustes para compensar pérdidas, y explicar cualquier discrepancia entre el tamaño muestral pretendido y el finalmente conseguido. No es particularmente interesante calcular a posteriori la potencia de un ensayo porque los intervalos de confianza (véase el punto 17) indican la imprecisión de las estimaciones32.
En algunos estudios se realizan análisis intermedios para decidir si vale la pena continuar con la inclusión de pacientes o, por el contrario, interrumpir el ensayo. Estos estudios requieren métodos de análisis que tengan en cuenta la multiplicidad de análisis33-35, y es necesario describir los que se han utilizado y el número de análisis realizados.
8. Generación de la secuencia aleatoria. En general, la asignación aleatoria se considera uno de los pilares fundamentales en que descansa la credibilidad de los resultados de un ensayo clínico (algunos autores preconizan un procedimiento de asignación, no necesariamente aleatorio, denominado minimización36). El término aleatorio tiene un significado técnico preciso que no siempre es bien comprendido37 y que a veces se utiliza con ligereza8. En ocasiones la descripción de los métodos de asignación utilizados no permite juzgar si esta asignación fue realmente aleatoria38. Por ello no basta con afirmar que la asignación fue aleatoria, sino que debe explicarse cómo se generó la secuencia de asignación (p. ej., mediante tablas de números aleatorios o mediante funciones generadoras de números aleatorios de programas informáticos), e indicar la razón de asignación (p. ej. 3:1) y las restricciones impuestas en el esquema de aleatorización (p. ej., ninguna, bloques de tamaño constante o variable, estratificación). Si se utilizó minimización, es necesario explicar cuáles fueron los factores de minimización y si se incluyó un componente aleatorio o fue enteramente determinista.
9. Asignación oculta. Para evitar que la asignación de los tratamientos sea sesgada no basta con generar una secuencia de asignación de tratamientos aleatoria39. La forma en que se utilice esta secuencia es crítica. Si quien ha de decidir la inclusión o no de un individuo en el ensayo conoce la secuencia de asignación de tratamientos, la decisión de inclusión puede estar condicionada (consciente o inconscientemente) por el tratamiento que recibirá el paciente en caso de que se lo incluya. El procedimiento de asignación debe garantizar que el tratamiento asignado es impredecible para quien decide la inclusión de individuos40, lo que se consigue manteniendo oculta la secuencia de asignación (concealed allocation)1. Para ello se puede utilizar distintos procedimientos, como la aleatorización telefónica por terceros o sistemas de asignación automatizados41. Dada la importancia del proceso de asignación de los tratamientos, es necesario describir el método utilizado.
10. Implementación. Para que se pueda juzgar si la asignación pudo ser predecible o no, además de describir el método utilizado, es particularmente importante explicar quién preparó la secuencia de asignación, quién decidió la inclusión de los individuos y quién realizó la asignación de los tratamientos.
11. Cegado (enmascaramiento). En un ensayo clínico, el término cegado, o a ciegas, se refiere a que los participantes ignoran qué tratamiento se ha asignado a cada individuo. Para conseguirlo, los distintos tratamientos deben enmascararse de forma que sean indistinguibles. Los ensayos sin enmascaramiento de los tratamientos se describen como ensayos abiertos.
El desconocimiento del tratamiento asignado por parte de quienes lo reciben, de quienes lo administran, de quienes evalúan el resultado42 y de quienes analizan los datos43 imposibilita que el prejuicio influya en el resultado del ensayo. El término doble cegado, muy utilizado en las publicaciones, es un tanto ambiguo: implica que quienes reciben y quienes administran el tratamiento ignoran cuál es éste, pero no revela lo que ocurre con los evaluadores y los estadísticos que analizan los datos44, extremo que debería quedar claro.
A diferencia de lo que ocurre con la asignación oculta (punto 9), el enmascaramiento de los tratamientos no siempre es posible. Aún así, en los ensayos abiertos, hay que justificar las razones por las que no se utilizó. En ocasiones, aunque los tratamientos se hayan enmascarado, sus efectos colaterales son demasiado reveladores. En estos casos, es importante realizar la evaluación a ciegas (lo que suele ser posible, incluso en ensayos abiertos, si el evaluador es una persona distinta de la que trata y controla al paciente) o verificar el éxito del enmascaramiento preguntando a los participantes (individuos que reciben y/o médicos que administran el tratamiento y evalúan el resultado) cuál creen que ha sido el tratamiento administrado45,46. Asimismo, la evaluación a ciegas es especialmente importante cuando las medidas utilizadas son subjetivas.
12. Métodos estadísticos. Los métodos estadísticos utilizados para analizar los datos deberían haberse predeterminado durante el diseño del estudio, y es importante indicar si realmente fue así, tanto para el análisis principal como para los secundarios, o bien si se decidieron tras disponer de los datos.
La mayoría de los métodos de análisis estadístico proporcionan una estimación puntual del efecto (entendido como una comparación de los tratamientos) y un intervalo de confianza (IC) para la estimación, que usualmente se determina con un nivel del 95%. El IC del 95%, que representa la incertidumbre sobre el efecto estimado, puede interpretarse como el conjunto de valores posibles que son compatibles con los datos del estudio.
Los resultados del análisis pueden expresarse también mediante un grado de significación (p), que expresa la probabilidad de los resultados observados (o más extremos) bajo la hipótesis nula de la correspondiente prueba estadística. Es preferible indicar los valores exactos (p. ej., p = 0,008) en lugar de utilizar ciertos puntos de corte (p. ej., p < 0,05)47.
Las pruebas estadísticas más usuales se basan en la suposición de que las observaciones son independientes. Cuando se dispone de múltiples observaciones para cada individuo (medidas en distintos tiempos o en distintos lugares del cuerpo), las distintas observaciones de un mismo individuo no son independientes, y es necesario utilizar estrategias o métodos de análisis que tengan en cuenta esta dependencia48-50. La dependencia de las observaciones puede ocurrir también en diseños en que los tratamientos se asignan a agregaciones (clusters) de individuos (como centros, médicos, colegios o unidades geográficas), pero las observaciones se realizan en cada individuo51.
Los análisis ajustados deberían incluir solamente variables de ajuste realmente importantes, y señalar si éstas se han preespecificado. En cualquier caso, la decisión de ajustar (o no) no debería basarse en la constatación (o no) de diferencias basales significativas52,53. En cuanto a los análisis de subgrupos, es más recomendable ajustar un modelo lineal que incluya una variable indicador del subgrupo y un término de interacción de ésta con el tratamiento que analizar cada subgrupo por separado54.
Resultados
13. Flujo de participantes. Para facilitar la comprensión del ensayo18, el flujo de participantes durante las distintas etapas (inclusión, seguimiento y análisis) debe ilustrarse mediante un diagrama como el de la figura 1. Idealmente debería indicarse el número de individuos inicialmente evaluados para su inclusión en el ensayo (y el de los que no se incluyeron, junto con los motivos) aunque este dato, que permite juzgar la validez externa de los resultados, no siempre está disponible. Es necesario indicar el número total de individuos aleatorizados y, para cada intervención, los números de individuos que se asignaron a la intervención, que la recibieron (tal como estaba previsto), que completaron el estudio según lo estipulado en el protocolo y que se incluyeron en el análisis principal, documentando las sucesivas exclusiones junto con los motivos. Esta información es crítica para juzgar la validez interna de un ECA, ya que las pérdidas y las exclusiones del análisis pueden introducir sesgos en los resultados, especialmente si se dan con diferente frecuencia en los distintos grupos55-57. El diagrama de la figura 1 puede requerir cierta adaptación según cuál sea la naturaleza de la intervención u otras características del ensayo23.
14. Reclutamiento. Las fechas en que se realizó el estudio son importantes para ubicarlo en su contexto histórico. Las fechas de inicio y final del reclutamiento permiten conocer su duración, dato que puede interesar a otros investigadores. En los ensayos con seguimiento de igual duración para todos los pacientes, la fecha de final del seguimiento permite conocer esta duración. En los ensayos en que la variable principal es el tiempo hasta un evento (p. ej., supervivencia), la duración del seguimiento no es la misma para todos los pacientes ya que el ensayo termina en una fecha concreta predeterminada. En estos casos, debe indicarse esta fecha, así como la duración mediana del seguimiento. Si el ensayo se interrumpió como resultado de un análisis intermedio (punto 7), debe indicarse34.
15. Datos basales. Las características demográficas y clínicas basales de los individuos incluidos en un ensayo son importantes para evaluar hasta qué punto los resultados de este ensayo pueden aplicarse a un paciente determinado. Estas características deben describirse para cada uno de los grupos y es recomendable hacerlo en una tabla. Las variables continuas pueden describirse mediante la media y la desviación estándar (mejor que errores estándar o intervalos de confianza) o mediante los tres cuartiles en caso de asimetría marcada. Las variables cualitativas y las cuantitativas discretas con un número limitado de valores pueden describirse mediante frecuencias. A pesar de que es una práctica común, no es útil realizar pruebas formales para comparar los grupos en cuanto a las características basales, y tales pruebas se han desaconsejado58,59. Aunque una asignación aleatoria correctamente implementada asegura la ausencia de sesgos de selección, no garantiza un equilibrio basal perfecto. Si ocurren desequilibrios, serán debidos a fluctuaciones aleatorias (no a sesgo) y las pruebas estadísticas que se utilizarán en el análisis principal sirven, precisamente, para establecer una conclusión teniendo en cuenta las posibles fluctuaciones del azar. Por tanto, asumiendo que la aleatorización es correcta, las decisiones de realizar análisis ajustados por las características basales (p. ej., análisis de covarianza) deben adoptarse en fase de diseño y no a resultas de constatar determinados desequilibrios en ellas53,60.
16. Números analizados. El número de casos incluidos en el análisis es un dato crítico para interpretar su resultado. Este número queda documentado en el diagrama de flujo para el análisis principal, pero puede ser distinto en otros análisis y debe quedar claro cuál fue en todos ellos. Para ello, es muy útil expresar los resultados de las variables dicotómicas en forma de fracción (p. ej., 10/20 en lugar de 50%).
En principio, para evaluar la efectividad de las intervenciones, es aconsejable realizar los análisis por intención de tratar (ITT), es decir, con la inclusión de todos los casos aleatorizados y manteniéndolos en el grupo al que se los asignó, porque así se evita posibles sesgos debidos a pérdidas o exclusiones no aleatorias61. Sin embargo, la adopción de esta estrategia no siempre es sencilla: si hay pérdidas de pacientes a lo largo del ensayo, la evaluación final prevista no estará disponible, lo que obligará a utilizar técnicas de sustitución de datos ausentes para mantener estrictamente la estrategia de análisis por ITT. Por otra parte, las desviaciones del protocolo en lo que se refiere a la administración de las intervenciones pueden inducir una subestimación del efecto, por lo que es recomendable realizar análisis adicionales que incluyan solamente los casos en que se administró la intervención tal como estaba previsto en el protocolo, lo que se denomina a veces análisis por protocolo (PP)62.
A diferencia de lo que ocurre cuando se desea demostrar diferencias entre las intervenciones, cuando el objetivo de un ensayo es demostrar la equivalencia de ambas o la no inferioridad de una respecto de otra, la estrategia por ITT no es conservadora: ciertas desviaciones del protocolo (notablemente las relativas a la administración de la intervención) pueden sesgar los resultados a favor de la hipótesis de equivalencia o no inferioridad63-65. Por otro lado, la potencia de un análisis PP será inferior a la del análisis por ITT66. Por esta razón, en estos ensayos es particularmente importante ofrecer el resultado de ambos análisis, ITT y PP, junto con las definiciones de los conjuntos de individuos que los integran para evitar usos abusivos de esos términos, en particular, el de ITT7.
17. Resultados y estimación. Todos los resultados (principal y secundarios) deben describirse para cada grupo de tratamiento (p. ej., medias y desviación estándar o frecuencias), junto con un estadístico adecuado que compare los resultados en ambos grupos (p. ej., diferencia de medias, diferencia de proporciones, riesgo relativo, odds ratio, NNT, etc.) y su IC. Aunque los IC se determinan casi siempre para una confianza del 95%, se puede utilizar otros niveles de confianza, pero deben haberse prefijado. Un error frecuente que debe evitarse es ofrecer los IC de los resultados en cada grupo en lugar de hacerlo para el estadístico que los compara. También puede ofrecerse el grado de significación (p) obtenido en una prueba estadística, pero además (no en lugar) del IC67,68.
Es importante facilitar el IC de todos los resultados, principales y secundarios, y no limitarse a los que reflejan diferencias estadísticamente significativas. Con ello se evitará tres fenómenos indeseables: el sesgo de publicación69,70, la errónea pero frecuente tendencia a considerar equivalentes intervenciones que no han mostrado diferencias estadísticamente significativas en ensayos infradimensionados71,72, y la tendencia a evaluar la significación estadística olvidando la relevancia clínica73.
18. Análisis auxiliares. Es inevitable que en un ECA convenga realizar más de un análisis inferencial. Sin embargo, la multiplicidad de estos análisis conlleva un elevado riesgo de resultados falsos positivos, por lo que conviene resistirse a la tentación de analizar demasiadas cosas, especialmente subgrupos de casos74,75, y concentrarse en las cuestiones realmente importantes y previstas en el protocolo o en el plan de análisis. Como ya se ha comentado en el punto anterior, es imprescindible informar acerca de todos los análisis realizados aunque puede ser innecesario ofrecer detalles de todos ellos.
19. Eventos adversos. Además del efecto pretendido, la mayoría de las intervenciones tienen efectos indeseados, y a veces indeseables. El equilibrio entre los beneficios y los riesgos es lo que debe juzgarse para decidir si una intervención es aceptable o útil. Por tanto, es imprescindible facilitar datos sobre los efectos indeseados. Al menos debe describirse la frecuencia de individuos que presentaron acontecimientos adversos (AA) serios (con una definición operativa de lo que se entiende por AA serio76), y de los que interrumpieron prematuramente el tratamiento (incluidos los motivos). Cuando un AA se ha presentado en más de una ocasión en el mismo individuo, es útil describir también la frecuencia de los AA.
20. Interpretación. Se ha señalado que, en las publicaciones científicas, la discusión de los resultados suele esmerarse más en justificarlos que en poner de manifiesto posibles limitaciones de estudio77,78. Para evitar esta tendencia, algunas revistas aconsejan estructurar la discusión79,80 y presentar: a) un breve resumen de los resultados clave, enfatizando lo que añaden al conocimiento previamente disponible; b) posibles mecanismos y explicaciones de los resultados; c) la comparación con otros trabajos similares, incluido un metaanálisis cuando sea posible; d) las limitaciones del estudio y los métodos adoptados para compensarlas; e) las implicaciones para futuras investigaciones, y f) las implicaciones clínicas.
21. Generalización. La validez externa del estudio, esto es, la posibilidad de generalizar los resultados a conjuntos de individuos más amplios que los incluidos en el ensayo, depende de diversos factores. En primer lugar, de la validez interna, que es un requisito previo para la validez externa: los resultados de un estudio sesgado son inválidos y ya no procede cuestionarse si son generalizables. Los criterios de selección, que usualmente hacen referencia a la edad, el sexo, la gravedad de la enfermedad, la comorbilidad y las comedicaciones, condicionan en gran medida la generalización a otros pacientes. Por ello es imprescindible plantearse seriamente la necesidad y la conveniencia de cada uno de ellos durante el diseño del ensayo, y no imponerlos a menos que exista un buen motivo, ya sea legal, ético o científico. La imitación de estudios previos no es, per se, un buen motivo. Además de los criterios de selección, muchos otros aspectos del ensayo determinarán también hasta qué punto pueden generalizarse sus resultados, como el ámbito en que se realizó el ensayo (atención primaria, pacientes hospitalizados, etc.), los métodos utilizados para diagnosticar la indicación del ensayo y para realizar el seguimiento, la intensidad del seguimiento, y la propia intervención o las condiciones en las que se administra. Posiblemente, se evitarían limitaciones en la validez externa de los ensayos si durante su diseño se tuviera muy presente la diferencia entre ensayos explicativos y ensayos pragmáticos81,82, y se decidiera explícitamente el objetivo principal en estos términos. En cualquier caso, decidir si los resultados de un ensayo son aplicables a un paciente concreto es un tarea tan difícil como importante para la práctica clínica83, sobre la que se han desarrollado guías84.
22. Evidencia global. Los resultados de un ensayo deben juzgarse en el contexto de la información previa disponible. Aunque lo ideal sería metaanalizar los resultados del ensayo junto con los de ensayos previos similares85, esto es poco práctico, muy costoso, y sólo factible si existen tales ensayos. El requisito mínimo es la discusión de los resultados del ensayo en el contexto de la evidencia disponible, sin limitarla a los estudios que obtuvieron resultados similares.

