


The simplest and most extended definition of myeloid-derived suppressor cells (MDSC) refers to them as immature myeloid cells with the ability to downregulate adaptative immune responses, a definition that reflects both their origin and function. Although initially described in experimental models and patients with cancer, accumulations of MDSC have also been found in other pathological conditions such as chronic/acute infections, autoimmune diseases and different types of stress. In all these situations MDSC may play their physiological role by modulating normal immune responses, both adaptive and innate. The mechanisms of action of MDSC are diverse requiring either cell to cell contact or the release of soluble factors. A better understanding of MDSC biology will open new windows of therapeutic opportunities, either by inhibiting their function (i.e. in cancer patients), or by enhancing their suppressive effects and promoting their expansion (i.e. in inflammation or autoimmunity).
La forma más común y sencilla de definir a las células supresoras de origen mieloide (MDSC, por sus siglas en inglés) resalta sus dos rasgos más característicos, dejando claro tanto su origen como su función. Aunque en un principio se identificaron en modelos experimentales y en pacientes con cáncer, con el tiempo se ha visto que en algunas enfermedades autoinmunes, infecciones (tanto agudas como crónicas) y diferentes tipos de estrés también se producen acumulaciones de estas células. En todas estas circunstancias, las MDSC contribuirían a limitar la intensidad de la respuesta inmune, tanto la innata como la adaptativa. Los mecanismos de acción de las MDSC son muy diversos: en ocasiones requieren el contacto intercelular, mientras que en otras circunstancias dependen de la presencia de factores solubles. Dado su papel inmunorregulador, numerosos estudios se han encaminado últimamente a determinar el potencial terapéutico tanto de su inhibición (p. ej., en pacientes con cáncer), como de la potenciación de su actividad (p. ej., en inflamación o autoinmunidad).
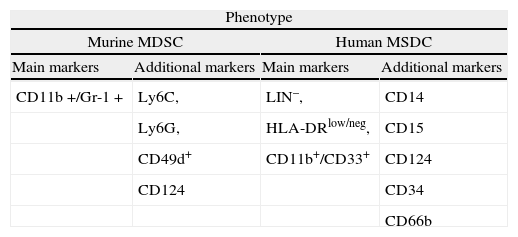
Accumulation of myeloid cells with suppressive activity on adaptative immune responses were described more than 20 years ago in tumor bearing mice1 and subsequently also in cancer patients. Since then, they were found to constitute a heterogeneous population of more or less immature myeloid cells that accumulate in the blood and lymphoid organs not only in cancer, but also in other pathologic situations2-6. The term MDSC was recently proposed7 and recent studies suggest that chronic inflammation and the expansion and activation of MDSC play a prominent role in tumor progression by downregulating antitumor immunity, mainly by impairing CD8+ T cell responses and NK-mediated cytotoxicity8-10. Given the wide range of cell types that may be included in this category, to find a phenotypic profile that characterizes all of them has been a difficult task (see Table 1). A consensus has been reached by using markers that unmistakably denote their myeloid origin: Gr-1 and Cd11b (Mac-1) in rodents and CD33 and CD11b (and negativity for lymphoid, NK, monocytic and DC markers) for human MDSC11.
Phenotype of MDSC and factors involved in their expansion
| Phenotype | |||
| Murine MDSC | Human MSDC | ||
| Main markers | Additional markers | Main markers | Additional markers |
| CD11b+/Gr-1+ | Ly6C, | LIN–, | CD14 |
| Ly6G, | HLA-DRlow/neg, | CD15 | |
| CD49d+ | CD11b+/CD33+ | CD124 | |
| CD124 | CD34 | ||
| CD66b | |||
| Expansion factors | |||
| Cyclooxygenase 2, prostaglandinsGranulocyte/macrophage colony stimulating factor (GM-CSF)Macrophage colony stimulating factor (M-CSF)Stem cell factor (SCF)Vascular endothelial growth factor (VEGF)IL-1b, IL-6, TGFbHypoxiaMicrobial related products (activators of toll-like receptors) |
Although in the majority of tumors and inflammatory models in rodents it is possible to find abnormal accumulations of MDSC, its presence is not necessarily detrimental. In normal mice, CD11b+Gr-1+ account for 20–30 % of bone marrow cells and 2–4 % of splenocytes, and are typically absent in the lymph nodes12. In healthy humans, immature myeloid cells represent about 0.5 % of the peripheral blood (PB) mononuclear cells13. All murine MDSC expresses the surface markers CD11b and Gr-1. CD11b is the a subunit of the β2 integrin Mac-1, which is expressed in granulocytes, dendritic cells (DC), monocytes and macrophages, and regulates leukocyte adhesion and migration. The Gr-1 antigen is detected by the RB6-8C5 antibody and is expressed from early myeloid committed progenitors in the murine bone marrow to mature cells in the PB14. RB6-8C5 antibody binds, though with different specificity, two phosphatidylinositol-anchored cell surface glycoproteins: Ly6C and Ly6G. Although Ly6C and Ly6G are predominantly expressed on the surface of monocytes and granulocytes, they can also be detected on endothelial cells, T lymphocytes and NK cells, among other cell types15. In addition to its broad use as a marker, recent evidence implicates the Ly6G molecule in regulating cell proliferation and apoptosis, possibly through the activation of a signal transduction cascade involving several STAT family members16.
The nuclear morphology and content of immunosuppressive substances have also been used to characterize murine MDSC. Morphologically, two major subpopulations have been described: granulocytic MDSC (G-MDSC) and monocytic MDSC (M-MDSC). G-MDSC have pseudosegmented or ring-shaped nuclei and contain high levels of arginase-1, while M-MDSC are mononuclear and contain both arginase-1 and iNOS12. Immunophenotypically, the expression level of the Gr-1 antigen has allowed the distinction of at least two cellular populations among CD11b+splenocytes isolated from tumor bearing mice: a Gr-1high, mainly composed of granulocytes in different stages of development, and a Gr-1int/low, comprising monocytes and other immature myeloid cells17. These two subpopulations differ in their ability to suppress T cell responses, although these differences may depend on the clinical or experimental setting. The use of specific antibodies recognizing Ly6C and Ly6G molecules revealed a similar distinction between M-MDSC, typically CD11b+Ly6G+/- Ly6C+, and G-MDSC, which have a CD11b+Ly6G+Ly6Clow phenotype. In general, Gr-1high cells are Ly6G+Ly6Clow SSCint granulocytes displaying weak or no suppressive activity, whereas Gr-1int/low cells are quite variable in their morphology and composition, as they contain a mixture of Ly6G+and Ly6G- cells, with some immature cells bearing ring-shaped nuclei that may express macrophage markers18. However, it has to be taken into account that when the Gr-1 antibody is used together with LyG6 specific antibodies, the intensity of the staining of the Ly6G antibody decreases whereas Ly6C staining seems unaffected19, which may lead to imprecise characterizations. In addition, binding of Gr-1 antibody to its epitopes in murine bone marrow cells was shown to signal via STAT-1, STAT-3 and STAT-5, similar to GM-CSF, and impair the immunosuppressive activity of MDSC16.
In addition to Gr-1 and CD11b, other markers have been used to characterize specific subsets of MDSC. In tumor-bearing mice, Gr-1+ CD11b+cells were shown to co-express the immature myeloid antigens CD31 and ER-MP5820,21. Other markers, possibly related to their suppressive function, activation or developmental stage include the co-stimulatory molecules CD80, CD40 and PD-L, the cytokine receptors CD115 (M-CSF-1R) and CD124 (a chain of IL-4 and IL-13 receptors), F4/80 or CD16/32. MDSC typically express MHC class I but low amounts of class II22-26, and do not express CD11c, as opposed to myeloid DC.
In a model of ovarian carcinoma, an expanded CD11b+ Gr-1+ cell population expressed CD80, as opposed to similar cells obtained form healthy mice22. Likewise, in mice with disseminated candidiasis, a population of CD11b+Gr-1+ CD80+ myeloid cells was found to be increased27. In both cases this costimulatory molecule was required for antigen specific immunosuppression. However, another study performed in tumor-bearing mice failed to identify a correlation between suppressive activity and expression levels of CD115, CD124, CD80, PD-L1 and PD-L218.
The lack of a definitive phenotype has fueled the search for alternative markers. Using a genomic approach, a recent study has identified CD49d as a molecule preferentially expressed in CD11b+Gr-1+ MDSC. In murine models of inflammatory bowel disease and cancer the use of this marker allowed differentiation of two distinct subpopulations among MDSC, a monocytic CD11b+Gr-1+ CD49d+subset that suppressed T cell proliferation (equivalent to the CD11b+Gr-1low/int cells), and the CD49d- fraction, morphologically granulocytic, that only poorly inhibited T cell responses28. These findings underline the phenotypic heterogeneity of MDSC. Because the expression of these markers is highly variable, aside from Gr-1 and CD11b, there are not unambiguous cell surface markers that define at present all mouse MDSC populations.
In addition to cancer, MDSC can be induced by chronic infections, sepsis, severe burn injury, traumatic stress and autoimmunity12. In all these situations, MDSC arise from myeloid progenitors under the influence of tumor-derived soluble factors (TDFs) or inflammatory mediators released during the processes. Consequently, the differences in phenotype and the range of cellular subpopulations present may depend on the specific combination of factors within the host. In an illustrative example, a model of colon carcinoma engineered to produce high amounts of GM-CSF induced the accumulation of myeloid intermediates that could be subdivided into two subsets on the basis of CD124 expression. CD124+ cells, morphologically resembling M-MDSC, were able to suppress T cell function, whereas CD124- cell fraction included granulocytic-like cells at various stages of differentiation that were not able to inhibit CD8+ T cell responses26. It is also important to note that these different subsets have been shown to have different suppressive effects in various clinical or experimental settings. For example, in CFA-treated mice, CD11b+Ly6C+MDSC inhibited OVA-specific proliferation by NO-mediated mechanisms, while the CD11b+Ly6G+cells had no suppressive capacity29. By contrast, in a mouse tumor model, both granulocytic and monocytic subsets were able to suppress OVA-specific responses, although using different mechanisms17. In the spleens of healthy mice, Greifenberg et al. distinguished up to six subpopulations based on size, granularity and the pattern of expression of CD11b and Gr-1. Gr-1high CD11blow (G-MDSC) as well as Gr-1low CD11bhigh Ly6Chigh SSClow (M-MDSC) populations functionally behaved as MDSC, inhibiting T cell responses, whereas Gr-1high CD11bhigh neutrophils and Gr-1low CD11bhish SSClow eosinophils were not suppressive. Interestingly, in this study, Gr-1high CD11blow cells were considered G-MDSC although they expressed F4/80, high levels of Ly6C and low levels of CD115, markers associated with monocytic cells. Similar distribution of myeloid suppressor subsets was found in the spleens of mice treated with a combination of LPS and IFN-y30. Later on, using a similar strategy, this group identified two populations with suppressor activity also in the bone marrow of healthy mice. They were both CD11b+but differed in the expression levels of Gr-1. CD11b+Gr-1high cells were morphologically similar to G-MDSC isolated from the spleen, whereas the CD11b+ Gr-1low subset was quite heterogeneous but contained mostly monocytic cells resembling M-MDSC16.
The absence of a Gr-1 homolog in humans has been an obstacle to identify human MDSC yet there is abundant information supporting the existence of both granulocytic and monocytic subsets. Human MDSC have been described mainly in cancer patients, but their phenotype is less well defined than in tumor-bearing mice. Both granulocytic and monocytic human MDSC express CD33, CD11b and CD124, and have variable expression of CD15 and other markers. M-MDSC are characterized by their additional expression of CD14 and lower levels of CD15 than their granulocytic counterparts. In addition, both subsets have low expression of HLA-DR molecules2,31. PBMC from renal cancer patients may contain a population of CD11b+CD14- CD15+ cells morphologically resembling G-MDSC and displaying increased arginase-1 activity. These cells mainly consisted of activated granulocytes expressing CD66b and VEGFR113,32. MDSC were also defined as CD11b+CD14- CD15+ CD33+ in the PB of patients with advanced non-small cell lung cancer33. More frequently, human MDSC have been identified in the monocytic fraction. In the PB of patients with melanoma, MDSC were defined as CD14+ CD11b+HLA-DRlow/neg. This population was undetectable in healthy donors34. MDSC with a similar phenotype were also reported in the PB of prostate cancer patients35. Likewise, CD14+/arginase+cells were described in patients with multiple myeloma and head and neck cancer. Also the PB of hepatocarcinoma patients contained a subpopulation of CD14+ HLA-DR- monocytes with suppressive activity. Importantly, the same population could be isolated from the tumor microenvironment36. In a recent study, specifically designed to test the immunosuppressive properties of different leukocyte subsets isolated from the PB of patients with melanoma and colon cancer, two main cell populations with the ability to suppress adaptative immune responses were identified, one expressing CD14 and the other CD15. Similar to murine MDSC, both populations expressed CD124 although only in the monocytic enriched fraction was the inhibitory activity correlated with expression of this marker37.
Despite the recent description of differentiated human monocytic and granulocytic subsets, earlier studies defined MDSC as a myeloid immature population comprised of monocytes and DC in an early stage of development. These non-activated MDSC could differentiate into granulocytes, monocytes or DC2. This immature signature is still a common finding. For instance, in the PB of renal carcinoma patients, MDSC are enriched in the fraction of cells negative for lineage markers (Lin-) and with low or absent expression of HLA-DR. The morphological analysis of this population revealed a heterogeneous mixture of monocytic and granulocytic cells38. In a recent study, Diaz-Montero et al. analyzed blood samples from patients with solid tumors to determine whether circulating MSDC levels correlated with the clinical stage of the tumor (I to IV). MDSC were defined as Linlow/neg HLA-DR- CD33+ CD11b+. Interestingly, among patients in Stage IV, higher percentages of MDSC were associated with extensive metastatic burden39. In agreement with the immature signature of MDSC, an increase in myeloid cells expressing CD34 was found in the blood and tumor infiltrates of patients with head and neck cancer. Also, a population of CD34+ CD33+ CD15- cells with suppressive ability was identified in the PB of patients with head and neck cancer and non-small cell lung cancer40,41.
Mechanisms of MDSC expansionIn cancer, factors inducing MDSC expansion include a variety of cytokines produced by tumor cells or tumor stromal cells which stimulate myelopoiesis and inhibit terminal differentiation of myeloid lineages. They include cyclooxygenase 2, prostaglandins42,43, granulocyte/macrophage colony stimulating factor (GM-CSF)42, vascular endothelial growth factor (VEGF)44, stem cell factor (SCF)45, IL-646 or macrophage CSF (M-CSF)47. Upon binding to their specific receptors, most of these cytokines activate the signal transducer and activator of transcription 3 (STAT-3) signaling pathways. This enhances cell survival and proliferation, reduces apoptosis and prevents full differentiation of myeloid progenitors into mature cells. More recently, C/EBPβ transcription factor was shown to be critical for the induction of MDSC suppressive activity48. Recruitment of MDSC to tumor sites is promoted by S100Ab and S100A9 proteins, which also induce MDSC cell maturation at the expense of DC49,50. Tumor microenvironment was found to influence both the differentiation and function of MDSC. In this study, MDSC with similar phenotype isolated from the tumor site and the secondary lymphoid organs of the same animal were compared. MDSC isolated from the spleen were able to suppress antigen-specific T cell responses but not non-specific reactions by using ROS. In contrast, MDSC isolated from the tumor inhibited both types of T cell responses by arginase-1 and iNOS-mediated mechanisms. Transfer experiments demonstrated that tumor environment caused upregulation of arginase-1 and iNOS, downregulating ROS in MDSC isolated from the spleen. Hypoxia, via induction of HIF-1α, was shown to be the factor responsible for the changes observed51.
In inflammatory diseases, expansion, recruitment and activation of these cells are induced by microbial products and factors produced by activated T cells. In sepsis, MDSC are expanded through Toll-like receptors (TLR) and MyD88 signaling52. Indeed, in addition to MDSC induction by tumors, these cells can be induced experimentally by treating normal mice with IFN-y and LPS, which results in iNOS induction and the release of NO. A recent report showed that cannabinoid receptors activation induced significant mobilization of MDSC in the lymphoid tissues of healthy mice53.
In vitro generation of MDSCDifferent types of MDSC can be generated by the culture of monocytes or hematopoietic progenitors in the presence of cytokines and growth factors46,54,55, or tumor-derived products such as tumor exosomes56 and conditioned media57,58. As an illustrative example, MDSC were generated from murine bone marrow cultures after exposure to high doses of GM-CSF for 4 days, but also using lower concentrations of the cytokine and maintaining the cells in culture for 10 days21. Different subsets of MDSCs including those with CD115+ Ly-6C+(monocytic) and CD115+ Ly-6C- (granulocytic) phenotypes can also be generated from mouse embryonic stem (ES) cells. These MDSC suppressed T-cell proliferation by IL-10 and NO production. In vivo, these cells prevented alloreactive T-cell-mediated graft-versus-host disease upon adoptive transfer47. We have found that culture conditions used in standard murine bone marrow retroviral transduction protocols (that include exposure to mIL-3 and mSCF) result in the generation of a heterogeneous population of immature myeloid cells, including a CD11b+Gr-1low cell population displaying suppressive activity in vitro (Gomez et al., manuscript in preparation). Currently, the similarities and differences between the different types of MDSC generated using different protocols and experimental conditions, and the relationship between MDSC and other types of myeloid regulatory cells such as tumor associated macrophages (TAM), alternatively activated macrophages or immature myeloid DC remain to be fully established.
MDSC activationAlthough experimentally it is possible to separate expansion and activation of MDSC, in practice the same array of factors that promote the accumulation of MDSC in the tissues or in the blood is also responsible for their activation. These factors include, but are not restricted to, soluble factors released by activated T cells or the tumor microenvironment and bacterial and viral products.
Early studies showed that NO59 and ROS60 production were IFNγ dependent. Blocking IFNγ by neutralizing antibodies or disrupting IFNγ pathway signalling by using STAT1 deficient mice abolishes MDSC-mediated T cell suppression by preventing the upregulation of effector enzymes17. However, MDSC from IFNγ receptor deficient mice are equally effective in suppressing T cell responses as MDSC isolated from wild type mice, putting into question the relevance of IFNγ in MDSC activation31. Activation of IL-4R signalling by IL-4 or Il-13 was shown to increase arginase-1 expression and activity61,62, but Stat6 deficiency blocked this induction, which is consistent with its role downstream of IL-4R63. Also the IL-13 mediated production of TGFp by MDSC cells in a sarcoma model was found to be dependent on Stat664. However, in a breast cancer model, IL-4Rα mediated signals did not seem to be critical in the activity of MDSC63. As in the case of IFN-γ and Stat1, the IL-4R-Stat6 axis may not be involved in all cases of tumor-induced immune suppression.
Other pro-inflammatory cytokines can also induce and activate MDSC. Mice bearing tumors secreting IL-1β developed higher levels of MDSC than their control mates injected with non-secreting cell lines65. In addition, these MDSC showed increased levels of ROS and enhanced suppressive activity in comparison with their controls. In inflammatory reactions, IL-6 acts downstream of IL-1β. Since MDSC express IL-6R but not IL-1R46,66 a direct effect of IL-1 on MDSC can be ruled out. It is then likely that the effects initially attributed to IL-1 could actually be induced by IL-6. IL-1 induces IL-10 production by MDSC and downregulates IL-12 production by macrophages. Experiments with TLR4-deficient mice demonstrated that these effects are mediated by signalling trough the LPS-TLR4 pathway. However TLR4 deficient mice still accumulate MDSC, indicating that MDSC can also be activated by TLR4 independent mechanisms67. Prostaglandin E2 (PGE2) and cycloxygenase-2 (COX-2) are inflammatory mediators produced by different tumors. PGE2 upregulates arginase-1 levels and suppressive activity in CD11b+CD14- CD15+ MDSC cells isolated from renal cancer patients3. Addition of COX-2 inhibitor to BM cell cultures prevented myeloid cell differentiation to MDSC, partially restoring cell phenotype and modulating arginase-1 expression in myeloid APCs58.
Mechanisms of suppression by MDSCThe most striking functional feature that defines MDSC is their ability to suppress T cell mediated immune responses. Accumulation and activation of MDSC are usually observed simultaneously, since they are generally induced by the same or similar mechanisms. Mechanisms mediating the suppressive activity include production of arginase-1, iNOS (which generates NO)68, generation of reactive oxygen species (ROS) 69 and peroxynitrite8, up-regulation of COX-2 and PGE270, induction of regulatory T cells24, production of TGFp71, stimulation of IL-10 and inhibition of IL-12 production by macrophages72, down-regulation of L-selectin by T cells73 and depletion of cysteine74. Expression of arginase-1 and NO deplete L-arginine, which is essential for T cell proliferation and function. NO also impairs T cell function by inhibiting JAK3 and STAT5 pathways, MHC class II expression and by inducing T cell apoptosis. Both tumor microenvironments and activated T cells produce factors contributing to this activation, which include ligands for TLRs, IFNγ, IL-4, IL-13, PGE2 and TGFp. These factors act by activating the STAT1, STAT6 or NF-kB signaling pathways in the MDSC. IFNγ activates STAT1 which upregulates arginase-1 and iNOS expression by MDSC. Both IL-4 and IL-13 can bind to IL-4Rα, which induces STAT6 activation and secondarily arginase-1 expression61.
An important issue of MDSC is its antigen-specific versus non-specific immunosuppressive effect, which is relevant for its potential use for tolerance induction. Several studies have reported that MDSC may produce antigen-specific suppression leading to tolerance75-78. MDSC can present antigenic peptides to T cells, but their low expression levels of class II MHC and co-stimulatory molecules do not favor T cell activation. In addition, MDSC have several mechanisms to actively suppress T cell responses. A paradigmatic example is the production of peroxynitrite, a powerful oxidant that was shown to induce nitration of cytotoxic (CD8+) T cell receptor and subsequent conformational changes that alter its peptide recognition ability, so that this T cell will no longer respond to this antigen, while maintaining its responsiveness to nonspecific stimuli8. More recently, in a series of elegant experiments, the same group demonstrated that incubation of Ag-specific CD8+ cells with peptide-loaded MDSC from tumor bearing mice not only precluded signaling downstream of TCR, but also prevented subsequent signaling from peptide-loaded dendritic cells. Using double TCR transgenic CD8+ cells, they showed that these MDSC induced tolerance to the peptide presented by MDSCs, whereas T cell response to the peptide specific to the other TCR was unaffected. Peptide-loaded MDSC caused nitration of the molecules on the surface of CD8+ cells, specifically at the site of physical interaction between MDSC and T cells76.
As different types of MDSC use different suppressive mechanisms, they may also play different roles in antigen-specific versus non-specific suppression. M-MDSC mediated suppression does not require cell-cell contact and is mediated by arginase-1, NO, PGE2 and cytokines. On the other hand, suppression by G-MDSC is mediated by ROS,18 requires physical contact with T cells and is more likely to induce tolerance8,11. In experiments carried out in our laboratory, we found that M-MDSC expressing an autoantigen (MOG40-55) were more suppressive in vitro than G-MDSC, but these G-MDSC were the only cell subpopulation that showed a therapeutic effect when injected to mice with MOG-induced autoimmune experimental encephalomyelitis (EAE), a murine model of multiple sclerosis. This effect was antigen-specific, as G-MDSC transduced with a control vector had a suppressive effect in vitro but not in vivo. It is likely that both nonspecific and antigen-specific suppression mechanisms coexist in different proportions depending on the setting and the MDSC subtype.
Clinical applications of MDSCMDSC offer two distinct opportunities for therapeutic intervention. Because in cancer patients, especially in those receiving immunotherapies, the effects of MDSC are frequently detrimental, intervention must be oriented to reduce the effects of MDSC. This can be achieved by destroying them, by terminal promoting myeloid differentiation, by inhibiting or reducing MDSC expansion, or by inhibiting their function. On the other hand, in inflammatory or autoimmune diseases, MDSC, either generated ex uiuo or directly induced in vivo, can be used for immunosuppressive or tolerogenic purposes.
MDSC differentiation to macrophages or DCs can be achieved by clinically approved agents such as vitamin D3 or derivatives, and vitamin A or derivatives like all-transretinoic acid (ATRA). Administration of ATRA improved the anti-tumor effect of cancer vaccines in tumor-bearing mice79. It also reduced the accumulation of MDSC and improved antigen-specific immune responses in patients with renal-cell carcinoma80. Blocking pathways that participate in MDSC expansion, such as those of VEGF and SCF, may also reduce MDSC accumulation. Indeed, treatment with sunitinib, a clinically approved tyrosine kinase inhibitor that blocks c-kit, VEGFR, PDGFR, Flt3, CSF-1 and RET pathways, reduced MDSC accumulation and prevented T cell anergy and Treg development in tumor bearing mice81, and it was associated with a reduction in the number of MDSC and a reversal of CD4+CD25hiFoxp3+ Treg cell elevation in patients with renal cell carcinoma82. Other strategies are aimed at inhibiting the suppressive effect of MDSC. Blocking PGE2 production by COX-2 inhibitors reduced the expansion of MDSC83 and inhibited tumor growth in tumor-bearing mice43. Finally, MDSC can be depleted using radiotherapy, monoclonal antibodies targeting myeloid markers or chemotherapeutic drugs. Indeed, treatment with gemcitabine or 5-fluorouracil was shown to deplete MDSC while preserving the numbers of T or NK cells, which enhanced antitumor immune responses in tumor-bearing mice84-86. Another chemotherapeutic agent, docetaxel, reduced tumor growth and improved immune responses in mice bearing mammary tumors. Additionally, docetaxel was shown to kill mannose receptor (MR)+ MDSC while preserving myeloid cells with a proinflammatory phenotype87.
Other strategies are aimed at blocking MDSC immunosuppressive function. A logical approach is to inhibit the two most important effector enzymes mediating these effects (arginase-1 and iNOS). Inhibitors of these two enzymes include nitroaspirines (NO-aspirines) and phosphodiesterase 5 inhibitors. NO-aspirin inhibited both iNOS and arginase activity in tumor-associated MDSC. Aspirin inhibited arginase, whereas iNOS was inhibited by the NO released by the drug88. NO-aspirin also has antioxidant activity and was shown to reduce the local production of peroxynitrites, known mediators of the immune suppressive effect. PDE5 inhibitors are already in clinical use for the treatment of pulmonary hypertension, cardiac hypertrophy or erectile dysfunction. One of these drugs, sildenafil, decreased IL-4Rα expression and subsequently downregulated both arginase-1 and iNOS in murine MDSC. In vivo, sildenafil significantly delayed tumor growth and increased immune responses in tumor bearing mice89. In humans, sildenafil also restored T cell proliferative responses in PBMC from patients with multiple myeloma or head and neck cancer89. Finally, inhibition of the production of PGE2 by celecoxib, a non-steroidal anti-inflammatory drug that blocks cyclooxygenase 2 (COX-2) activity, prevented chemical induction of large intestinal tumors in association with a reduction in arginase-1 and iNOS expression and in the number of MDSC in the spleen of tumor bearing mice, together with a restoration of CD4+ cell numbers and functionality83.
The immunosuppressive and tolerogenic effects of MDSC may also be used for therapeutic purposes. Theoretically, since MDSC can behave as APC, they can be used to induce tolerance in antigen-specific settings. In this regard, monocytic CD11b+CD115+ Gr-1+ cells were found to participate in tolerance induction by co-stimulatory blockade with CD40L specific mAb in a murine model of cardiac transplantation77. Tolerance induction by MDSC was also reported in rodent models of allogeneic skin90 and kidney91 transplantation.
Final remarksAlthough the first observations reporting a negative role of a myeloid component in the antitumor responses were dated in the 80's, the last decade has lived an implosion of reports documenting their involvement in a number of different situations, the variety of their mechanisms of action and phenotypic diversity. Still, a number of questions remain unanswered6. MDSC definition excludes specifically any mention to a particular phenotype. The multiple of cell types that may be included in this category may simply translate the idea that immature myeloid cells can hardy accomplish their task in a functional immune response. In this context, an accumulation of immature myeloid cells in the periphery or tissues can be taken as an alarm sign of a situation that drives their expansion and their phenotype should be a reflection of the pathological circumstances that induce such an increase in numbers (Table 1). Their role as regulators of the immune responses makes them as an interesting target for therapeutic intervention. In our view, with the presently available therapeutic tools, the possibility of reducing their number or function in cancer patients seems more plausible than trying to expand or activate them in cases of overreactivity of the immune system. Without doubt more effort has to be invested before ensuring MDSC are not just another cell population revisited by the immunology community, but expectation is great.
Conflict of interestsThe work in the authors's laboratory has been financed by grants from the Spanish MICINN, the Instituto de Salud Carlos III, and the European Union. Ramon Gimeno and Jordi Barquinero are researchers of the Miguel Servet program.