



Las células dendríticas (CD) juegan un papel fundamental en la regulación de la respuesta inmune. Son las principales células presentadoras antigénicas, por su capacidad de capturar, procesar y presentar antígenos de forma óptima a linfocitos T, y generar respuestas inmunes específicas. Posteriormente al descubrimiento de esta función y al aparecer técnicas metodológicas que permitían su purificación y maduración in vitro, se ha comprobado que también son capaces de activar otros tipos celulares, como linfocitos B, células NK, macrófagos o eosinófilos, e incluso generar tolerancia inmunológica. Este mejor conocimiento de su biología y funciones ha permitido el desarrollo de ensayos clínicos basados en el uso de CD en el campo de la inmunoterapia antitumoral y antiinfecciosa o para inducir tolerancia postrasplante o en patologías autoinmunes.
Dendritic cells (DC) play a critical role in the regulation of the immune response. They are the main antigen presenting cells, due to their ability to capture, process and present antigens to T lymphocytes in an optimal way, and to generate specific immune responses. Subsequently, when methodological techniques allowed their in vitro purification and maturation, it was verified that they were able to activate several other immune cell subsets, like B lymphocytes, NK cells, macrophages or eosinophils, and even to induce immune tolerance. A deeper knowledge of their biology and functions has allowed the development of clinical trials in anti-tumour and anti-infective DC-based therapies, and also to induce post-transplant tolerance and to treat autoimmunity.
El hallazgo del papel central de las células dendríticas en la respuesta inmune parte de observaciones de Cohn et al. y Steinman et al.1,2. Estos autores, mediante variadas técnicas de separación celular, eran capaces de enriquecer una población de células procedentes de tejidos linfoides que parecían mucho más eficaces que los macrófagos promoviendo la proliferación de linfocitos T aloreactivos3. Hasta ese momento, se pensaba que los macrófagos eran las más potentes células presentadoras antigénicas profesionales.
Los conocimientos biológicos sobre este nuevo tipo celular avanzaron lentamente en la década de los 80, debido a la complejidad metodológica derivada de la purificación de una población minoritaria de los tejidos linfoides. Este panorama cambió radicalmente en los 90, ya que una serie de descubrimientos metodológicos permitieron la diferenciación in vitro de células que compartían marcadores celulares y características funcionales con las descritas CD4–7. En el año 2000 se caracterizaron una serie de antígenos específicos de las CD sanguíneas, los Blood Dendritic Cell Antigens (BCDA)8 Desde que estas técnicas de identificación y diferenciación in vitro fueron disponibles, se avanzó rápidamente en el conocimiento de las CD.
En las dos últimas décadas se han hecho espectaculares progresos en el entendimiento del papel que ejercen las CD en el sistema inmune, derivados de los múltiples y variados estudios en diferentes aspectos del ciclo celular de las mismas. Entre ellos se incluyen los mecanismos de captura, procesamiento y presentación de antígenos derivados de patógenos, células apoptóticas o tumorales, la sinapsis inmune entre las CD y los linfocitos T, el descubrimiento de receptores lectina tipo C en la captura antigénica, el control de la activación de las CD a través de receptores Toll-like (TLR), la importancia de las CD plasmacitoides en la respuesta antiviral, el papel de la CD en la polarización de la respuesta de linfocitos T helper, la regulación de las respuestas de linfocitos B y células natural killer (NK), el análisis in vivo del tráfico de las CD y las interacciones inmunes, el control de inmunidad versus tolerancia por las CD, el papel de las CD en las infecciones microbianas, patologías autoinmunes, reacciones alérgicas, rechazo de trasplantes, y respuestas inmunes antitumorales, y las vías de diferenciación de las CD.
Estos avances en la inmunobiología de las CD, junto con las técnicas de transfección y diferenciación in vitro, han permitido el desarrollo de prometedoras terapias en el campo de la inmunoterapia antitumoral y antiinfecciosa, así como para promover tolerancia postrasplante o en enfermedades autoinmunes.
Características de las CDLas CD maduras (CDm) presentan una morfología propia, caracterizada por la presencia de numerosos procesos membranosos que pueden tomar la forma de dendritas, pseudópodos o velos. Contienen altas concentraciones de estructuras intracelulares relacionadas con el procesamiento antigénico, como endosomas, lisosomas o los gránulos de Birbeck de las células de Langerhans de la epidermis. Están presentes en tejidos y órganos linfoides y no linfoides, así como circulantes en linfa aferente y sangre periférica. Reciben diferentes nombres según la ubicación, pero guardan características y funciones similares entre si.
Fuera de tejidos linfoides, son abundantes en piel, faringe, esófago alto, vagina, ectocérvix y ano, y en las superficies mucosas de los sistemas respiratorio y gastrointestinal9. Extienden sus procesos membranosos entre las estrechas uniones de las células epiteliales sin alterar la función de la barrera epitelial10. Esto aumenta la captura de antígenos del entorno incluso si no hay infección o inflamación, conduciendo al silenciamiento del sistema inmune ante los antígenos ambientales inocuos.
Aparecen en las regiones T dependientes de los ganglios linfáticos y bazo, donde se las conoce como células interdigitantes. En el bazo son más numerosas, ya que hay nidos de ellas en la periferia del área de linfocitos T, donde están posicionadas como puentes a través de los cuales deben pasar los linfocitos para entrar en el torrente sanguíneo. Las células dendríticas foliculares se encuentran en los centros germinales de los folículos secundarios de las áreas de linfocitos B de ganglios linfáticos y bazo, siendo parte integral del microambiente del folículo. También están presentes en el timo, sobre todo en la región medular. En linfa aferente se las conoce como células veladas, representando células de Langerhans migrantes en tránsito desde la piel al ganglio linfático donde se transformarán en células interdigitantes. En sangre periférica constituyen menos del 2% de las células mononucleares.
También se encuentran en corazón, hígado, parénquima pulmonar y lámina propia del intestino. En el cerebro no se han descrito; sin embargo, las células de la microglía se asemejan a CD por la forma y por sus marcadores de membrana.
Un obstáculo para la detección y estudio de las CD hasta el descubrimiento de los BDCA ha sido la ausencia de un marcador en la superficie celular específico de esta estirpe celular. Se caracterizan por la elevada expresión de antígenos MHC clase II y la ausencia de marcadores de linaje como CD14 (monocitos), CD3 (linfocitos T), CD19, CD20 y CD24 (linfocitos B), CD56 (células NK) y CD56b (granulocitos)11. Presentan moléculas de adhesión comunes con monocitos y macrófagos (CD11a, CD11c, CD50, CD54, CD58, CD102)12 y moléculas coestimuladoras (CD40, CD80, CD86)13,14. Su fenotipo varía a lo largo de los diferentes estados de maduración y activación. Los precursores circulantes en la sangre pueden expresar CD2, CD4, CD13, CD16, CD32 y CD33, que van perdiendo gradualmente con la maduración15. Por el contrario, las moléculas coestimuladoras, de adhesión y los antígenos del MHC aumentan a lo largo de la maduración. Otras moléculas, como CD80 y CD86, incrementan su expresión con la activación, principalmente tras la unión de CD40 a su ligando. CD86 es un marcador temprano de maduración, mientras que CD80 aparece más tardíamente y está ausente en los precursores circulantes16.
Subclases de CDCon la excepción de las CD ganglionares foliculares, que son de origen mesenquimal17, las CD se originan a partir de progenitores hematopoyéticos medulares CD34+ por la influencia del ligando de FLT-3 (sFlt-3L) y GM-CSF18. Estos precursores CD34+ se diferencian en células progenitoras comunes mieloides (CMP) y células progenitoras comunes linfoides (CLP).
En sangre periférica circulan dos tipos de CD fenotípica y funcionalmente diferentes. Se distinguen por su expresión de CD11c y CD12319,8,20. Las CD CD11c+CD123lo tienen una morfología monocitoide, por lo que se denominan CD monocitoides o mieloides (MDC), mientras que las CD CD11c-CD123hi presentan características morfológicas similares a las células plasmáticas, por lo que se han denominado CD plasmacitoides (PDC). Se diferencian a partir de CMP y CLP, respectivamente. En humanos, las MDC pueden derivar de precursores ya comprometidos hacia CD o bien de precursores del linaje de granulocitos y monocitos. También pueden originarse a partir de diversos tipos celulares previamente a su diferenciación definitiva; precursores de monocitos y granulocitos pueden diferenciarse hacia CD cuando se exponen in vitro a combinaciones apropiadas de citoquinas, que incluyan GM-CSF y TNFα en presencia o ausencia de IL-421,22. PDC y MDC difieren en numerosos aspectos, incluyendo su distribución tisular, su producción de citoquinas y los factores de crecimiento necesarios para su diferenciación.
Las MDC se pueden dividir en dos subpoblaciones según su expresión de BDCA1 (CD1c) y CD141: MDC1 (células Lin−CD4+HLA DR+CD11c+CD1c+CD141−) y MDC2 (células Lin−CD4+HLA DR+CD11c+CD1c-CD141+)8. También se clasifican según su localización: residentes en tejidos periféricos, residentes en órganos linfoides secundarios y MDC circulantes. Estas últimas en humanos sanos suponen un total de 14,2±3,9células/μl20. Las mejor estudiadas son las MDC de la piel, localización en la que al menos se pueden diferenciar tres tipos de MDC: las células de Langerhans (CLs), que residen en la epidermis, y DC CD1a+ y DC CD14+, que aparecen en la dermis. Las DC CD14+ expresan un gran número de lectinas tipo C, como DC-SIGN, DEC-205, LOX-1, CLEC-6, Dectin-1 y DCIR. En contraste, las CL expresan las lectinas langerina y DCIR. Estos dos subtipos celulares también difieren en el panel de receptores TLR que presentan: las MDC CD14+ dérmicas presentan TLR2, 4, 5, 6, 8 y 10, mientras que las CL expresan TLR1, 2, 3, 6 y 1023–25. Otra discrepancia entre estos dos subtipos celulares es el tipo de citoquinas que secretan. Mientras que las DCs CD14+ producen una gran variedad de factores solubles, como IL-1β, IL-6, IL-8, IL-10, IL-12, GM-CSF, MCP y TGFβ, las CL secretan una pequeña cantidad de citoquinas, entre las que destaca la IL-1526. Esta producción de citoquinas diferencial explica las diferentes funciones biológicas de los distintos subtipos de MDC. Las MDC CD14+ dérmicas inducen la diferenciación de los linfocitos T CD4+ naive hacia un subset de linfocitos T CD4+ especializados en la cooperación con los linfocitos B, activando su producción de grandes cantidades de IgM y el cambio isotópico hacia IgG e IgA. De estas funciones es responsable la IL-21 producida por los linfocitos T CD4+, inducida a su vez por la IL-12 secretada por las MDCs CD14+ dérmicas27. Por el contrario, las LC son eficientes inductoras de respuesta de linfocitos T CD8+, parece ser que por la acción de IL-1528.
Las PDC son importantes en las respuestas inmunes antivirales y en procesos autoinmunes. Constituyen la principal fuente de INF I tras las infecciones29. Circulan por el torrente sanguíneo y entran en los órganos linfoides a través de las vénulas de endotelio alto. En sujetos sanos suponen 8,9±1,7células/μl20. Estas células, marcadores de linaje−HLA DR+CD11c−, expresan altos niveles de CD123 y marcadores específicos como BDCA-2 (CD303) y BDCA-4 (CD304)30. A diferencia de las MDC, presentan TLR1, 6, 7, 9 y 1029. Pueden ser activadas por virus31, IL-3 y ligando de CD40 de origen mastocítico32, y por componentes microbianos como motivos CpG de ADN33. Por su enorme plasticidad funcional, son capaces de desencadenar diferentes respuestas inmunológicas. Cuando son expuestas a virus viables, inician respuestas de memoria, induciendo la expansión y diferenciación de linfocitos B de memoria antígeno específicos a células plasmáticas33, y de linfocitos T antígeno específicos, a CTL34. Por el contrario, las PDC activadas por motivos CpG o IL3/CD40L inducen la secreción de IL-10 por parte de linfocitos T CD4+ reguladores35 y la activación de linfocitos T CD8+ supresores por la expresión del ligando de ICOS36.
Se ha comprobado la existencia de dos subtipos de PDC según su expresión de CD2. Ambos subtipos se han encontrado tanto circulantes como en amígdala; sin embargo, las PDC CD2high también se detectan en algunas biopsias tumorales, sugiriendo alguna función en la inmunovigilancia tumoral. Tanto las PDC CD2high como las PDC CD2low secretan IFNα y expresan las moléculas citotóxicas granzima B y TRAIL. Las PDC CD2high son potentes activadoras de la respuesta de linfocitos T. En contraste, las PDC CD2low parecen presentar una capacidad limitada para inducir la proliferación de linfocitos T alogénicos. Estas diferencias funcionales entre los dos subtipos de PDC están asociadas con distintos patrones de transcripción, secreción diferencial de IL12p40 y expresión diferencial de la molécula coestimuladora CD80 tras su activación37.
En humanos, las PDC pueden ser movilizadas in vivo con citoquinas como el ligando de Flt3 y GM-CSF, mientras que las MDC son movilizadas únicamente por Flt318,38. La movilización diferencial de diferentes subclases o precursores de CD por estas citoquinas ofrece una nueva estrategia de manipular las respuestas inmunes en humanos.
Maduración de las CDSe han propuesto dos modelos para explicar la diferenciación de las CD a partir de precursores hematopoyéticos. Uno postula un único linaje de precursores de CD dotado de plasticidad funcional, mientras que el otro defiende la existencia de varios linajes de precursores de CD funcionalmente diferentes39. Ambos modelos definen tres estados de maduración: precursores de CD, CD inmaduras (CDi) y CD maduras (CDm). Para comprender las funciones de las CD, deben de entenderse sus diferentes estadíos de maduración.
Los precursores de CD migran de la médula ósea y circulan por la sangre hasta lugares específicos del organismo donde maduran y actúan como centinelas del sistema inmune. Este tráfico hacia los tejidos periféricos está dirigido por la expresión de los receptores de quimioquinas CCR1, CCR5 y CCR6, y por moléculas de adhesión como el ligando de CD62P40–42.
Las CDi expresan CCR1 y CCR3, cuyos ligandos se expresan en los endotelios activados y en células inflamatorias, promoviendo su migración hacia piel, bazo, hígado, pulmón, corazón, riñón y mucosas, constituyendo una red de CD intersticiales en todos los órganos excepto en sistema nervios central y testículo11. La función principal de las CDi es la captura antigénica. Las CDi procesan antígenos adquiridos tanto endógenamente (sintetizados en el citosol de las CD) como exógenamente (procedentes del entorno extracelular). Los posibles orígenes de antígenos extracelulares incluyen bacterias, virus, células necróticas o apoptóticas, proteínas de choque térmico, inmunocomplejos y otros tipos de proteínas. Son capturados por diversos mecanismos (macropicnocitosis, endocitosis mediada por receptor y fagocitosis) gracias a los receptores de superficie presentes en las CD, como receptores de Fc43, integrinas44, lectinas tipo C45, y los llamados receptores scavenger, como LOX-1 y CD9146.
Ya en el interior de las CD, los antígenos proteicos son degradados a péptidos que se unirán a las moléculas del complejo principal de histocompatibilidad de clase I (MHC I) o clase II (MHC II) y serán transportados a la superficie celular para el posterior reconocimiento por linfocitos T antígeno-específicos. Los antígenos proteicos endógenos, procesados a través de la vía del MHC I, son primeramente ubiquitinados y degradados en péptidos por el proteosoma en el citosol. Posteriormente son transportados al retículo endoplásmico (RE) por moléculas transportadoras asociadas con el procesamiento antigénico (TAP), donde son cargados en el MHC I. Los complejos MHC I-péptido son llevados desde el RE a la membrana celular a través de la red trans-Golgi para la presentación a los linfocitos T CD8+.
Los antígenos proteicos exógenos son procesados en endosomas que se fusionan con lisosomas portadores de proteasas, responsables de la degradación de las proteínas en péptidos que se unirán a moléculas de MCH II. Los complejos MHC II-péptido son transportados por túbulos especializados a la superficie celular, donde se presentarán a linfocitos T CD4+. Estas proteínas exógenas también pueden ser procesadas por las CD a través del MHC I, fenómeno conocido como «presentación cruzada»; esto permite la activación de una respuesta inmune tanto de linfocitos CD4+ como de linfocitos T CD8+ ante antígenos exógenos47.
Los antígenos lipídicos, tanto los expresados en patógenos (por ejemplo, micolatos de micobacterias) como los presentes en tejidos propios (esfingolípidos, fosfatidil inosítidos), son presentados por las CD a través de la molécula CD1, que heterodimeriza con la β2-microglobulina48. El procesamiento de estos antígenos lipídicos hasta las diferentes moléculas CD1(a-d) se realiza en compartimentos intracelulares especializados. El repertorio celular restringido por CD1d incluye linfocitos T con su característica diversidad en el TCR, así como células NKT invariantes.
El fenotipo de las CDi se caracteriza por la expresión de grandes cantidades de receptores de quimioquinas inflamatorias, que les permiten extravasarse a los tejidos inflamados49,50 y por una baja expresión de moléculas co-estimuladoras, de adhesión, de MHC y marcadores específicos de CD.
La maduración de las CD es un complejo proceso que conduce a la diferenciación final de las CD, transformándolas desde unas células escasamente inmunoestimuladoras que funcionan como centinelas periféricos que capturan antígenos, en las más potentes células estimuladoras de linfocitos T. Además de la captura de antígenos, las CD deben recibir las llamadas «señales de peligro» originadas por patógenos, inflamación o daño tisular51,52 para sufrir este proceso madurativo que les permitirá desplazarse a los órganos linfoides secundarios y estimular de forma eficiente los linfocitos vírgenes. Estas señales madurativas se deben tanto a moléculas inflamatorias originadas en el propio organismo (ligando de CD40, TNFα, IL-1, IL-6, IFNα) como a productos microbianos o bacterianos agonistas de los TLR.
Con la maduración las CD adquieren una gran motilidad y pierden su capacidad de capturar antígenos al disminuir la expresión de receptores de fagocitosis y endocitosis. Las CDm optimizan el procesamiento de antígenos aumentando la expresión de los componentes de la maquinaria enzimática responsable del proceso53,54, y adquieren la capacidad de presentar antígenos y estimular a los linfocitos T tras el incremento en la expresión de moléculas del MHC y de moléculas de adhesión y co-estimulación (CD40, CD54, CD58, CD80, CD83, CD86). La mayoría de estos marcadores ya están presentes en bajos niveles en las CDi; en cambio, el CD83 está ausente en las CDi y sirve como marcador para distinguir las CDi de las CDm. Algunas de estas moléculas están implicadas en la señalización bidireccional entre la CD y el linfocito T, modulando tanto la activación del linfocito como las funciones de la CD.
Durante la maduración, las CD disminuyen la expresión de los receptores de quimioquinas CCR1 y CCR5, y aumentan la de CCR7. El ligando de este último se encuentra en las paredes de los ganglios linfáticos y en la zona paracortical ganglionar55. Las propias CD secretan quimioquinas como TARC, MDC o IP-10, que reclutan diferentes tipos de linfocitos T, y RANTES, MIP-1α y MIP-1β, que reclutan monocitos y otras CD hacia el microambiente local.
Funciones de las CDLas CD estimulan a los linfocitos T de una manera mucho más potente que los macrófagos o los linfocitos B. Su expresión de moléculas de MHC es entre 10 y 100 veces mayor que la de los linfocitos B3. Imágenes a tiempo real de CD murinas y linfocitos T naive en ganglios linfáticos intactos revelan que una CD puede interaccionar hasta con 500 linfocitos T en una hora56,57.
La activación eficaz de los linfocitos T por parte de las CD necesita de varias señales consecutivas. Las CD pueden activar tanto a linfocitos T CD4+ como linfocitos T CD8+ por presentación antigénica vía MHC clase II y MHC clase I, respectivamente, lo que constituiría la primera señal. La segunda señal se realiza por la interacción con moléculas coestimuladoras presentes en las CDm: CD80 y CD86 con el receptor linfocitario CD28, y la familia TNF con los receptores linfocitarios R-TNF. Si falla esta coestimulación, los linfocitos T se vuelven tolerogénicos.
Tras su activación, los linfocitos T vírgenes sufren una expansión clonal y una diferenciación a células efectoras secretoras de citoquinas y células memoria. El tipo de respuesta consiguiente de los linfocitos T depende de varios factores, como la concentración antigénica en la CD, la afinidad del TCR por el MHC, la duración de la interacción de la CD con el linfocito T, el estado de maduración de la CD, y el tipo de estímulo responsable de la maduración de la CD58. La supervivencia a largo plazo de los linfocitos T y su diferenciación a células de memoria y efectoras requiere la interacción con CD maduras. La activación inducida por CD inmaduras es de más corta duración.
Los linfocitos CD4+ pueden diferenciarse hacia linfocitos Th-1, que producen IFNγ y secundan una respuesta de linfocitos T CD8+ citotóxicos, o hacia linfocitos Th-2, que producen IL-4, IL-5 e IL-13, e implican inmunidad humoral y disminución de la respuesta Th-1. El patrón de citoquinas producido por la CD activada determinará la dirección de esta polarización; IL-12, IL-18 e IL-27 dirigen hacia Th-1, mientras que CCL17, CCL22 o la ausencia de IL-12 lo hacen hacia Th-2. A su vez, este patrón de citoquinas está regulado por factores como el subtipo de CD, la localización anatómica de la CD o el tipo de estímulo madurador. Estos parámetros también controlan otras características de la respuesta de los linfocitos T, como la inducción de tolerancia o la recirculación (homing) de los linfocitos T.
La cooperación de los linfocitos T CD4+ en el momento de la activación es necesaria para generar linfocitos T CD8+ memoria. Se cree que esta interacción está mediada por la unión entre la molécula CD40 de la CD y su ligando en el linfocito CD4 activado, el CD40L59, aunque hay estudios que sugieren una interacción directa entre los linfocitos CD4+ y el CD40 de los linfocitos CD8+60. Otras moléculas que se han implicado en la generación de células de memoria y en las respuestas duraderas son proteínas pertenecientes a las familias de CD28 y del receptor de TNF (TNFR).
Además de su papel central en la activación de los linfocitos T, las CD interactúan directamente con células NK, células NKT y linfocitos B. CDm y CDi pueden activar e inducir la expansión de células NK resting por mecanismos no aclarados totalmente, aunque se han descrito algunos proteínas y factores solubles implicados61,62. Las CD activadas también inducen directamente la proliferación de linfocitos B, el cambio de isotipo de inmunoglobulinas y su diferenciación a células plasmáticas secretoras de anticuerpos; estas acciones las pueden llevar a cabo tanto de forma linfocito T-dependiente como linfocito T-independiente.
Es reseñable el papel de la CD en la generación de la tolerancia inmunológica antígeno-específica en el control de los fenómenos autoinmunes. Las CD tímicas promueven la eliminación de los linfocitos T autorreactivos63, y las CD periféricas inducen tolerancia principalmente en su estado inmaduro o semimaduro64. Como ya se ha comentado, la presentación antigénica en ausencia de moléculas coestimuladoras o sin IL-12 inducen linfocitos T reguladores que suprimen la respuesta inmune mediante la secreción de IL-10 y TGFβ. Las propias CD sufren un proceso de parada madurativa y se vuelven tolerogénicas en presencia de sustancias como esteroides, vitamina D3, IL-10, TGFβ 65,66 o CTLA-4 producido por la población reguladora CD4+CD25+FoxP3+67.
Obtención de precursores de CDDesde lo primeros estudios en los que se aislaban CD del bazo de ratones, se han desarrollado diferentes protocolos para optimizar la obtención de una cantidad adecuada de CD que permita su uso clínico, principalmente en protocolos de inmunoterapia tumoral. Inicialmente se obtenían precursores de CD presentes en sangre periférica mediante centrifugación en gradiente de densidad, pero el proceso no era demasiado rentable dada su escasa frecuencia. En los últimos años se han conseguido obtener del orden de 1x106-1x108 CD mediante técnicas de cultivo ex vivo.
Una de las técnicas más empleadas en la diferenciación de CD a partir de monocitos de sangre periférica. Los monocitos se purifican de productos de aféresis por elutriación, inmunoselección o adherencia. Posteriormente se diferencian a CD en presencia de GM-CSF e IL-4, y maduran con TNF, IL-1β, PGE2 o IL-6, en un protocolo que tiene una duración de 6-10 días21,68.
También se pueden usar células CD34+ como fuente de CD. En este caso, las células se obtienen a partir de productos de aféresis, médula ósea o cordón umbilical mediante inmunoselección y se cultivan con GM-CSF y TNFα durante 6-10 días69,70.
Otra alternativa es la expansión de CD in vivo. El uso de citoquinas como G-CSF71 o Flt3L72,73 aumenta considerablemente el número de precursores de CD en sangre periférica, que pueden ser recogidos mediante una aféresis y purificados mediante inmunoselección. Esta estrategia acorta el tiempo de cultivo ex vivo, ya que estos precursores maduran rápidamente, en 24-48 horas.
Algunos grupos han hecho comparaciones de las CD obtenidas a partir de diferentes técnicas. Cuando se comparan CD de sangre periférica con CD obtenidas a partir de monocitos, se observa que las primeras fenotípicamente parecen estar más activadas, ya que precisan de un menor periodo de tiempo para madurar, mientras que en las segundas ese periodo de tiempo es muy variable y depende mucho del estímulo utilizado. Sin embargo, parece que las CD de sangre periférica son peores productoras de citoquinas, menos activas en endocitosis y carecen de la expresión de DC-SIGN, pero son mejores iniciadoras de respuestas T antígeno-específicas y presentan una mayor capacidad de polarizar la respuesta hacia un perfil Th171–73. Si se comparan CD derivadas de monocitos con CD derivadas de progenitores CD34+, ambos tipos presentan morfología y fenotipo similares, aunque la inducción de respuesta T antígeno-específica está aumentada cuando se utilizan las CD derivadas de progenitores CD34+74–76. Otros autores han publicado ensayos estudiando las diferencias entre distintas estrategias de diferenciación de CD a partir de monocitos, variando las citoquinas utilizadas tanto en su generación como en su maduración; los resultados presentados en este sentido son muchas veces contradictorios.
La inducción de una respuesta anti-tumoral exitosa requiere el uso de CD maduras inmunoestimuladoras por dos motivos: por su capacidad para inducir una respuesta T antígeno-específica y porque las CD inmaduras parecen inducir tolerancia inmune. Se ha comprobado que las CDi inducen la expansión de linfocitos T reguladores, aunque también se ha descrito que las CDm también pueden inducir esta expansión tras ciertos estímulos77–79. Además, se ha comprobado que las CDm son resistentes a ciertos factores inmunosupresores producidos por las células tumorales, y son fenotípica y funcionalmente estables en ausencia de citoquinas. En la tabla 1 se resumen los principales estímulos utilizados en la maduración de las CD.
Principales estímulos utilizados en la maduración de las CD
| Mediadores pro-inflamatorios | Interleuquinas (IL-1β, IL-6), interferones (IFNα/β, IFNγ), MCM, TNFα, PGE2 |
| Agonistas de moléculas co-estimuladoras | CD40L, OX40L, 4-IBBL |
| Agonistas de TLR | Peptidoglicano, zymosan, LPS, MPL, Ribomunyl®, Luivac®, Biostim®, OK432, caTLR4, flagelina, poli(I:C), Ampligen®, ssRNA, R848, poli-G10, oligonucleótidos CpG |
| Quimioquinas | CCL16 |
| Otros | Factor del complemento C5b-9, inmunocomplejos |
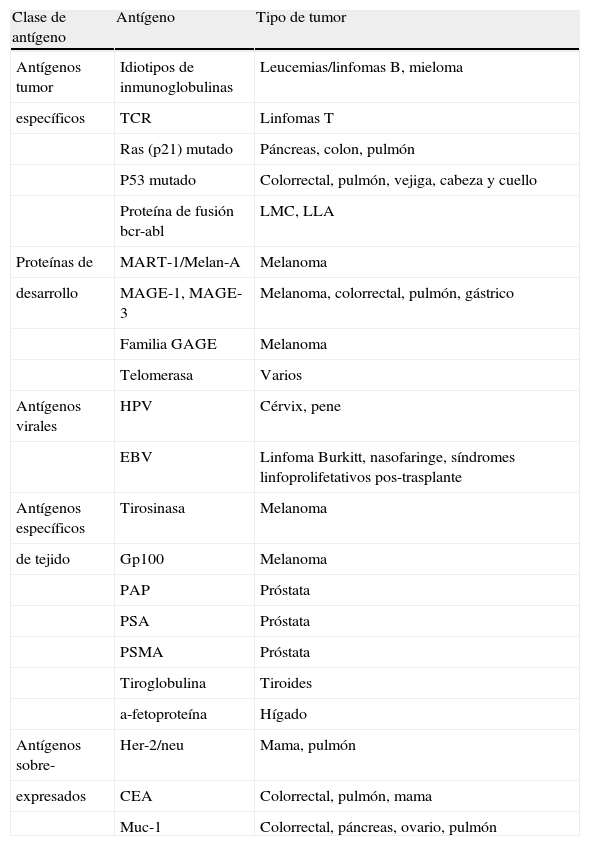
La capacidad de las CD para generar respuestas antitumorales in vivo ha sido documentada en modelos animales y en estudios clínicos humanos. La mayoría de los ensayos implican el aislamiento de CD, seguido de la carga con antígenos tumorales y la posterior infusión de estas CD portadoras de antígenos. Se han descrito un elevado número de antígenos tumorales susceptibles de ser utilizados en protocolos de inmunoterapia. Los más significativos se resumen en la tabla 2.
Antígenos tumorales potencialmente utilizables en inmunoterapia
| Clase de antígeno | Antígeno | Tipo de tumor |
| Antígenos tumor | Idiotipos de inmunoglobulinas | Leucemias/linfomas B, mieloma |
| específicos | TCR | Linfomas T |
| Ras (p21) mutado | Páncreas, colon, pulmón | |
| P53 mutado | Colorrectal, pulmón, vejiga, cabeza y cuello | |
| Proteína de fusión bcr-abl | LMC, LLA | |
| Proteínas de | MART-1/Melan-A | Melanoma |
| desarrollo | MAGE-1, MAGE-3 | Melanoma, colorrectal, pulmón, gástrico |
| Familia GAGE | Melanoma | |
| Telomerasa | Varios | |
| Antígenos virales | HPV | Cérvix, pene |
| EBV | Linfoma Burkitt, nasofaringe, síndromes linfoprolifetativos pos-trasplante | |
| Antígenos específicos | Tirosinasa | Melanoma |
| de tejido | Gp100 | Melanoma |
| PAP | Próstata | |
| PSA | Próstata | |
| PSMA | Próstata | |
| Tiroglobulina | Tiroides | |
| a-fetoproteína | Hígado | |
| Antígenos sobre- | Her-2/neu | Mama, pulmón |
| expresados | CEA | Colorrectal, pulmón, mama |
| Muc-1 | Colorrectal, páncreas, ovario, pulmón |
CEA: antígeno carcinoembrionario; EBV: virus de Epstein Barr; HPV: virus del papiloma humano; LLA: leucemia linfoblástica aguda; LMC: leucemia mieloide crónica; PAP: fosfatasa ácida prostática; PSA: antígeno prostático específico; PSMA: antígeno prostático específico de membrana; TCR: receptor de células T.
La exposición al antígeno puede hacerse siguiendo diferentes estrategias. La más frecuente es la incubación de las CD con péptidos derivados de proteínas tumorales restringidos por MHC y con epítopos definidos para la estimulación de linfocitos T, principalmente CD8+. Desde finales de la década de los 90 se han hecho ensayos utilizando péptidos derivados de antígenos tumorales como MART-1, MAGE-1, gp100, CEA, PSMA o HER-2/neu, por citar algunos80–82. Esta metodología se basa en algoritmos predictivos que identifican péptidos con alta afinidad por la molécula de HLA, más comúnmente con los alelos HLA*A0201. Este sistema tiene las ventajas de evitar procesos autoinmunes y el no requerimiento de células o tejido tumoral. Sus principales inconvenientes son la necesidad de conocer los epítopos tumorales y la posibilidad de aplicarla únicamente a los pacientes cuyo HLA sea el adecuado. Para evitar los mecanismos de escape tumoral, es frecuente el uso de varios péptidos simultáneamente, en lugar de un único péptido.
En caso de que el epítopo no esté definido, se suele utilizar la proteína tumoral completa. De esta forma se evita la restricción por un HLA concreto. Se han utilizado diferentes métodos para introducir las proteínas solubles en interior de las CD, como microinyección83, fusión mediada por liposomas84, electroporación85, o a través de su unión a otras moléculas que faciliten su transporte, como pueden ser fragmentos de receptor de Fc86, toxinas bacterianas87 o proteínas víricas88.
Con el objeto de intentar inducir una respuesta dirigida contra el mayor número de antígenos tumorales posibles, se han desarrollado estrategias en las que se cargan las CD con antígenos derivados de las células tumorales completas; esto también puede reducir la posibilidad del escape tumoral que puede ocurrir si la vacunación se hace con un pequeño número de antígenos. La congelación repetida o la sonicación de las células tumorales producen extractos que pueden aplicarse en este tipo de protocolos89,90. Otra opción es el uso de células tumorales a las que previamente se ha inducido apoptosis91 o necrosis92. Una estrategia más reciente emplea la fusión de las CD con las células tumorales mediante técnicas de electroporación93 o polietilenglicol94.
Por último, existen técnicas en las que se introduce ARN95,96 o ADN97,98 de las células tumorales en las CD, de forma que se consigue la expresión de los antígenos tumorales dentro de la CD y su posterior presentación en la membrana unidos al MHC.
Cada uno de los sistemas expuestos presenta ventajas e inconvenientes característicos, incluso se han obtenido resultados contradictorios utilizando el mismo método.
ConclusiónDesde su descubrimiento, las CD se han confirmado como las principales células presentadoras de antígenos, desempeñando un papel central en la respuesta inmune. Por ello constituyen la base de los intentos de conseguir un tratamiento inmunoterápico antitumoral eficaz. Durante décadas la generalización de protocolos en esta dirección ha estado dificultada por numerosos problemas metodológicos basados en la escasa información sobre su biología y funciones. En la actualidad se disponen de las técnicas y los conocimientos necesarios para introducir las terapias basadas en CD dentro del arsenal de la oncología moderna, así como en el tratamiento de otras patologías de origen infeccioso o inmunológico.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.