



Different polymorphisms in key genes of innate and adaptive immunity disrupt the intestinal host-microbiotic balance in inflammatory bowel disease (IBD). In most cases these alterations affect the maintenance of the intestinal epithelium. Because of this, actions taken in order to reinforce or help intestinal epithelium recovery can provide benefits to idiopathic illnesses such as IBD. With this in mind, several studies support the idea that actions derived from the Toll-like receptor (TLR) 2 of the epithelial cells may help to maintain the epithelial barrier. However, in inflammatory conditions TLR-2 disappears from the intestinal epithelium and acquires a pivotal role in dendritic cells. In this event, the interleukin (IL)-10 production by sub-epithelial dendritic cells mediated by TLR-2 activation, thus leading to T-reg phenotypes in the lamina propria, may favour the recovery from epithelial damage. Therefore, the TLR-2/IL-10 axis can help to reinforce and recover the epithelial lining in IBD. In this way, several data suggest that early epithelial repair diminishes the antigen load reaching the lamina propria, which reduces inflammation and improves therapeutic performance. However, there is currently a lack of knowledge regarding molecular regulation of epithelial barrier by TLR-2 and IL-10.
Distintos polimorfismos en genes clave de la inmunidad innata y/o adquirida son capaces de romper el equilibrio homeostático entre el huésped y sus bacterias comensales en la enfermedad inflamatoria intestinal (EII). Estas alteraciones genéticas pueden afectar al mantenimiento de la integridad del epitelio intestinal. Por lo tanto, el desarrollo de nuevas aproximaciones terapéuticas destinadas a fortalecer y a recuperar el epitelio intestinal lesionado podría ser beneficioso para estos pacientes. En este sentido, distintos estudios han constatado que la activación del receptor Toll-like 2 (TLR-2) en las células epiteliales puede ayudar a mantener la barrera epitelial. No obstante, durante la inflamación el TLR-2 desaparece del epitelio y adquiere protagonismo en las células dendríticas del compartimiento subepitelial de la mucosa intestinal. Éstas son capaces de producir interleucina (IL) 10 por activación del TLR-2, lo cual promueve la polarización la respuesta inmune hacia un fenotipo T-regulador en la lamina propria, lo que a su vez puede contribuir a la recuperación del epitelio dañado. En consecuencia, la activación del eje TLR-2/IL-10 puede ayudar a reparar la línea epitelial en la EII. En otros estudios se ha indicado que una pronta regeneración del epitelio intestinal supone una disminución de la carga antigénica que alcanza la lamina propria, lo que disminuiría la inflamación y facilitaría la respuesta terapéutica. A pesar de ello, el conocimiento de las bases moleculares que regulan las acciones de TLR-2 y IL-10 en el epitelio intestinal, y sus consecuencias en la EII, es todavía escaso.
Over the past decade, the study of intestinal mucosal immunity has been raised, mainly due to its relation to potentially severe and chronic illnesses with a growing incidence. This is the case of idiopathic affections such as inflammatory bowel disease (IBD), celiac disease or irritable bowel syndrome, amongst other of infectious nature (human immunodeficiency virus, enteropathogen infections, etc.). Crohn's disease and ulcerative colitis are two entities grouped under the label of IBD. Basically, these are chronic inflammatory illnesses with flare ups separated by intervals in which the disease is clinically inactive (although sub-clinical inflammation may persist, as often occurs in Crohn's disease). The midand long-term consequences of that are the deterioration of the patient's health and quality of life. In spite than the aetiology of IBD is still unknown, recently more than 30 genetic alterations which can predispose to suffer from IBD have been described. One of the first genes associated to Crohn's disease susceptibility, the nucleotide-binding oligomerization domain containing 2 (NOD2)/caspase recruitment domain member 15 (CARD15), is directly involved in the recognition of components from bacteria walls1. Three specific mutation forms of NOD2 (Arg702Trp, Gly908Arg, and a frameshift deletion mutation at Leu1007) are present in some 10 %-20 % of Caucasian patients with Crohn's disease. Studies of association in large series of patients have described other polymorphisms potentially associated with susceptibility of suffering from IBD (ATG16L1, IRGM, LRRK2, HLA-DRA, DRB1*0103, ICOSLG, IL12B, IL23R, MST1, OCTN1 and OCTN2, MDR1, PXR, MUC19, DLG5, CCRG, STAT3, IBD5, PTPN2, IL-10R, ARPC2, PTGER4, HNF4A, CDH1 and CDH3, LAMB1, IL-27, etc). These data reinforce the polygenic complexity of these diseases2-6. These mutations alter different tolerogenic mechanisms which regulate the interaction of the host with the intestinal microbiota, triggering a chronic inflammatory process which results in bowel damage. Therefore, imbalances in intestinal ecology grant a pathological role to bacteria. In any case, genetic and environmental components converge in IBD which provoke phenotypic and subphenotypic clinical variability, as well as its chronic course, and the development of local and systemic complications.
Many of the known TLRs agonists
| Microbial ligands | Synthetic ligands | Endogenous ligands | |
| TLR-1a | Tri-acyl lipopeptides, lipoarabionomannan, N. meningitides soluble factors | Tri-palmitoylated lipopeptide (Pam3CSK4) | |
| TLR-2 | Peptidoglycan, zymosan, glycoinositolphospholipids, atypical LPS, porins | Urate, hialuronan, Hsp60, Hsp70, Hsp96, HMGB1, apoliprotein CIII, oxidised LDL, serum amyloid A, amyloid beta, versican | |
| TLR-3 | dsRNA, poli I:C, CMVb | mRNA | |
| TLR-4 | LPS, viral envelope glycoproteins | Urate, fibrinogen, fibronectin EDA, b-defensin, surfactant protein A, Tamm-Horsfall protein, hyaluronan, heparin sulphate, Hsp60, Hsp70, Hsp96, HMGB1, Minimally-modified LDL, Oxidised LDL | |
| TLR-5 | Flagelin | ||
| TLR-6a | Di-acyl lipopeptides, lipotheichoic acid (LTA) Group B Strep heat-labile soluble factor, Staph phenol-soluble modulin | ||
| TLR-7 | ssRNA | Imidazoquinolinesc, loxoribine and bropirimine | RNA/protein complexesd |
| TLR-8 | ssRNA | Imidazoquinolinesc | |
| TLR-9 | Hypomethylated CpG DNA HSV-2 | IgG-chromatin complexes | |
| TLR-10 | Unknown | ||
| TLR-11 | Profilin |
TLR-10 is only present in humans and TLR-11 in mice. Recognition of ligands by dimerization with: a) TLR1:TLR2 or TLR2:TLR6; b) TLR3:TLR9; c) TLR7:TLR8; d) TLR7:TLR9.
Although different cellular functions are included under the denomination of intestinal barrier, the gut-associated lymphoid tissue (GALT) system is the main component involved in the defence against a wide and diverse repertoire of luminal antigens. GALT holds a high level of plasticity which allows it to respond to changes in intestinal microbiota thus maintaining immunological tolerance. The anticipated control of potential pathogens has been one of the evolutive advantages acquired by the immunological systems of vertebrates. An example of this model is the generation of a diverse repertoire of B and T cell antigen receptors through the rearrangement of immunoglobulin (Ig) V, D, and J gene fragments7. However, these systems are not free from errors produced by mutations, which are deleterious in some cases, but they occasionally show adaptive advantages such as interleukin (IL)-23 receptor Arg381Gln variant that protects non-Jewish individuals from suffering from Crohn's disease8. Indeed, a point genetic alteration may even be hidden by compensatory mechanisms to maintain the homeostasis. However, IBD patients accumulate several mutations in the GALT system which consequently, leads to unbalanced immune responses where the inflammatory ones predominate9.
The host-bacteria symbiosis plays a fundamental role in the development and maintenance of the intestinal mucosal integrity, as well as in increasing immunological tolerance. In particular, the epithelial cell line is a border which keeps the commensal bacteria away and establishes communication with mucosal immune cells by means of lymphokines 10. Intestinal epithelial cells do that job in different, sometimes complementary, ways. For instance, the mucins segregated by goblet cells increase their efficiency in the presence of bactericidal substances such as Paneth cells-derived defensins, cathelicidins and C-type lectins11,12. These glycoproteins and antimicrobial peptides are the natural unspecific mucosal defences in most living beings. Mammals are also equipped with more specialized defensive molecules which are also secreted into the intestinal lumen. This is the case of inducible polymeric-IgA which is produced in great quantities in the Peyer's patches by plasma cells13. The importance of this supra-epithelial defensive system has been proven in several studies which have shown that abnormalities in mucin functions or a decrease in IgA production diminish the mucosa protection and increase the risk of intestinal inflammation, in mice as well as in humans14-19.
On the other hand, NOD2 is expressed in antigen-presenting cells as well as in Paneth cells located at the base of ileal crypts. In this sense, Wehkamp et al. showed a significantly diminished expression of human a-defensins 5 and 6 in the involved ileum from Crohn's disease patients with NOD2 mutations20,21. The Wnt signalling pathway transcription factor-4 orchestrates Paneth cell differentiation and regulates the expression of Human a-defensins 5 and 6. Recently, the newly identified association of transcription factor-4-deficiency with ileal disease provides additional evidence that the decrease in Paneth cell a-defensins could be a primary factor in Crohn's disease pathogenesis22.
The main cellular mechanism involved in the control of inflammation in the intestinal mucosa is the apoptosis. Even in normal physiological conditions, the susceptibility of the CD3+CD4+ lamina propria lymphocytes to apoptosis was higher in healthy mice (wild type) than in IL-10 knockout mice with moderate spontaneous intestinal inflammation23. On the other hand, different studies have shown that the main mechanism associated to clinical effectiveness of both anti-tumor necrosis factor (TNF) or thiopurine therapies is the induction of apoptosis in macrophages and lymphocytes of the intestinal lamina propria24-27. In Crohn's disease there is increased T cell proliferation induced by undefined stimuli derived from bacterial antigens, food, and possibly unrecognised pathogens. At the same time, the lifespan of these cells is greater because of their resistance to apoptosis. This defective apoptosis results in inappropriate T cell accumulation that promotes bowel inflammation in Crohn's disease28. A recent work by our research team in active Crohn's disease patients treated with corticosteroids (namely, 1mg/kg/day prednisolone) found a significant increase in apoptosis of lamina propria T cells (CD3+) and B cells (CD19+) in those patients who responded to therapy as compared to those who where unresponsive to it24. However, the apoptotic index of these cells at baseline (before starting steroid therapy) did not differ between responders and non-responders24 suggesting that apoptosis resistance of CD3+ and CD19+ lamina propria lymphocytes is not a previously programmed mechanism, but an epiphenomenon which appears during inflammation. Also, studies in vitro suggest that steroids induce an apoptotic response in CD3+CD4+ cells, which varies in intensity depending of the activating stimulus29.
While the importance of decreased apoptosis of intestinal lymphoid cells in the pathogenesis and therapeutic response in IBD seems to be quite clear, the role of apoptosis of the epithelial cells is still being elucidated. The epithelial cells are generated from progenitor cells in the base of the crypt increasing its differentiation, until reaching the apical extreme where they die by physiologic apoptosis (anoikis). During this highly dynamic process, cross-talk mechanisms between epithelial cells (tight junctions, desmosomes and adherent unions), or with other surrounding cells (intraepithelial lymphocytes, natural killer cells, myofibroblasts, macrophages and dendritic cells) are established30, in order to maintain the intestinal homeostasis. Given this context, it is essential that the functional and proliferative epithelium be preserved. If not, the appearance of an excessive apoptosis in epithelial cells would cause intestinal barrier disruption. In fact, some studies have found early micro-erosions resulting from increased epithelial apoptosis in ulcerative colitis 31, while TNF-a promoted epithelial damage in Crohn's disease32.
Nuclear factor (NF)-ĸB is the most relevant transcription factor for the maintenance of the homeostasis of the epithelium, which consists in homodimers or heterodimers of different NF-kB/Rel proteins in mammals33. NF-ĸB is activated by proinflammatory cytokines such as TNF-α and IL-1β as well as a broad set of microbial or host-derived ligands through the Toll-like receptor (TLR) downstream34 (fig. 1). NF-kB activates transcription of numerous target genes, many of which encode cytokines and adhesion molecules that orchestrate the activation of inflammatory leukocyte subsets resulting in intestinal damage35. Colonic biopsies as well as lamina propria macrophages from IBD patients showed increased NF-ĸB activity and enhanced p65/RelA expression36,37. Moreover, a polymorphism in the promoter region of the NF-ĸB1 gene has been associated with an increased risk of developing IBD 38. Several studies have showed how inhibitor of ĸB kinase (IKK) complex/NF-kB canonical pathway inhibition in macrophages/neutrophils cells but not in epithelial cells ameliorated disease in mouse models of colitis39-41. However, epithelial cell-IKK ablation led to apoptosis of colonic epithelial cells, impaired expression of antimicrobial peptides and enhanced bacteria translocation into the mucosa19, generating a spontaneous IBD-like phenotype in intestinal epithelial cell-specific NEMO (IKK-g)-deficient mice.
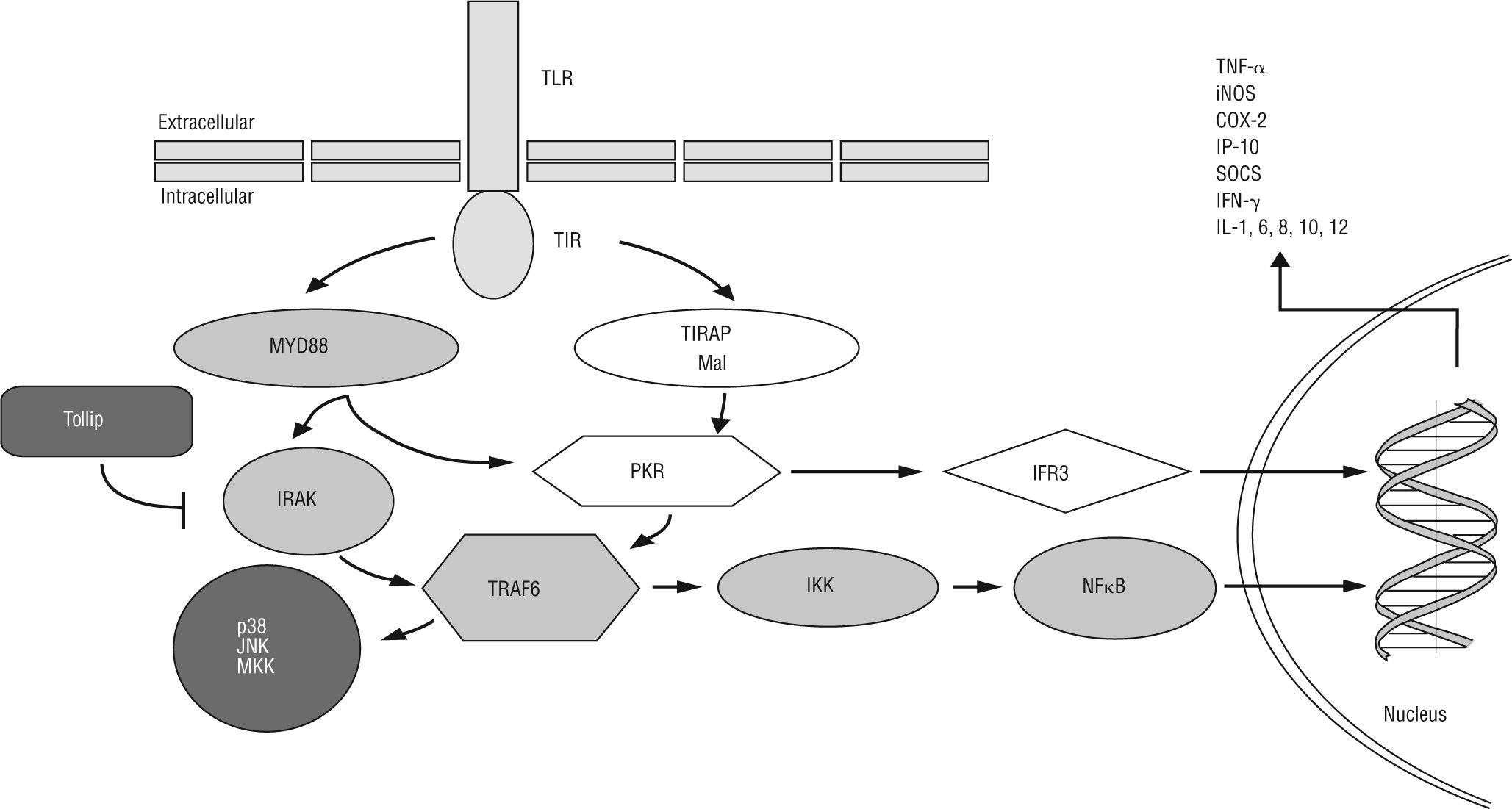
![TLRs are capable of differentially activating at least two distinct downstream signalling pathways after recognition of a pathogen-specific molecular pattern: a) MyD88-dependent TLR signalling pathway is activated via cytoplasmic Toll-interleukin 1 receptor (TIR), which provides a scaffold for recruitment of the adaptor molecule MyD88 and serine/threonine kinases of the IL-1R-associated kinase (IRAK) family. IRAK-activated TNF receptor-associated factor 6 (TRAF6)/IĸB kinase (IKK) axis induces NF-ĸB translocation to the nucleus, resulting in transcriptional activation of genes encoding cytokines and chemokines [e.g. TNF-α, inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), suppressor of cytokine signalling (SOCS), IFN-γ-induced protein 10 (IP-10), IFN-β and IL-1, 6, 8, 10, 12]. In addition, TLRs bridge the signalling via TRAF6 for mitogen-activated protein kinase (MAPK) kinase (MKK), p38 and jun N-terminal kinase (JNK) in response to specific bacterial products. Toll-interacting protein (Tollip) inhibits TLR2/4-mediated cell activation by suppressing the activity of IRAK. b) The MyD88-independent TLR signalling pathway activates the TIR-domain-containing adaptor protein (TIRAP), resulting in dsRNA-binding protein kinase (PKR) activation. This protein could mediate potential crosstalk in the TIRAP- and MyD88-dependent signalling pathways. The MyD88-independent pathway activates both IFN-regulatory factor 3 (IRF3) and NF-ĸB, and results in the expression of IFN-γ-inducible genes including IP-10.](https://static.elsevier.es/multimedia/02139626/0000003000000001/v1_201305141316/S0213962611700099/v1_201305141316/es/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNdDfbi4GraT4jwPl5dTud2oD7cjrpHNJVcuqElrgVT865LyaXCCc3jphZVV6G1gqofcU6ctb+t2795+rgDt1o9ds1pNzASbZ1gCmYOT6i5u+GOjAL51Na301slLKdrQbskir6G22xYIcCkUwQWJwExsY8KabS8bZLNSHGcpfD4fUrqxWAjyRzL2CGqEUYdUcV4ksUckvGjrgZ0d0A3YrNnUetO+LTeKyI0vTjRpSlzeAblIG+Pl5m8kpTukRzbFFD4= "TLRs are capable of differentially activating at least two distinct downstream signalling pathways after recognition of a pathogen-specific molecular pattern: a) MyD88-dependent TLR signalling pathway is activated via cytoplasmic Toll-interleukin 1 receptor (TIR), which provides a scaffold for recruitment of the adaptor molecule MyD88 and serine/threonine kinases of the IL-1R-associated kinase (IRAK) family. IRAK-activated TNF receptor-associated factor 6 (TRAF6)/IĸB kinase (IKK) axis induces NF-ĸB translocation to the nucleus, resulting in transcriptional activation of genes encoding cytokines and chemokines [e.g. TNF-α, inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), suppressor of cytokine signalling (SOCS), IFN-γ-induced protein 10 (IP-10), IFN-β and IL-1, 6, 8, 10, 12]. In addition, TLRs bridge the signalling via TRAF6 for mitogen-activated protein kinase (MAPK) kinase (MKK), p38 and jun N-terminal kinase (JNK) in response to specific bacterial products. Toll-interacting protein (Tollip) inhibits TLR2/4-mediated cell activation by suppressing the activity of IRAK. b) The MyD88-independent TLR signalling pathway activates the TIR-domain-containing adaptor protein (TIRAP), resulting in dsRNA-binding protein kinase (PKR) activation. This protein could mediate potential crosstalk in the TIRAP- and MyD88-dependent signalling pathways. The MyD88-independent pathway activates both IFN-regulatory factor 3 (IRF3) and NF-ĸB, and results in the expression of IFN-γ-inducible genes including IP-10.")
TLRs are capable of differentially activating at least two distinct downstream signalling pathways after recognition of a pathogen-specific molecular pattern: a) MyD88-dependent TLR signalling pathway is activated via cytoplasmic Toll-interleukin 1 receptor (TIR), which provides a scaffold for recruitment of the adaptor molecule MyD88 and serine/threonine kinases of the IL-1R-associated kinase (IRAK) family. IRAK-activated TNF receptor-associated factor 6 (TRAF6)/IĸB kinase (IKK) axis induces NF-ĸB translocation to the nucleus, resulting in transcriptional activation of genes encoding cytokines and chemokines [e.g. TNF-α, inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), suppressor of cytokine signalling (SOCS), IFN-γ-induced protein 10 (IP-10), IFN-β and IL-1, 6, 8, 10, 12]. In addition, TLRs bridge the signalling via TRAF6 for mitogen-activated protein kinase (MAPK) kinase (MKK), p38 and jun N-terminal kinase (JNK) in response to specific bacterial products. Toll-interacting protein (Tollip) inhibits TLR2/4-mediated cell activation by suppressing the activity of IRAK. b) The MyD88-independent TLR signalling pathway activates the TIR-domain-containing adaptor protein (TIRAP), resulting in dsRNA-binding protein kinase (PKR) activation. This protein could mediate potential crosstalk in the TIRAP- and MyD88-dependent signalling pathways. The MyD88-independent pathway activates both IFN-regulatory factor 3 (IRF3) and NF-ĸB, and results in the expression of IFN-γ-inducible genes including IP-10.
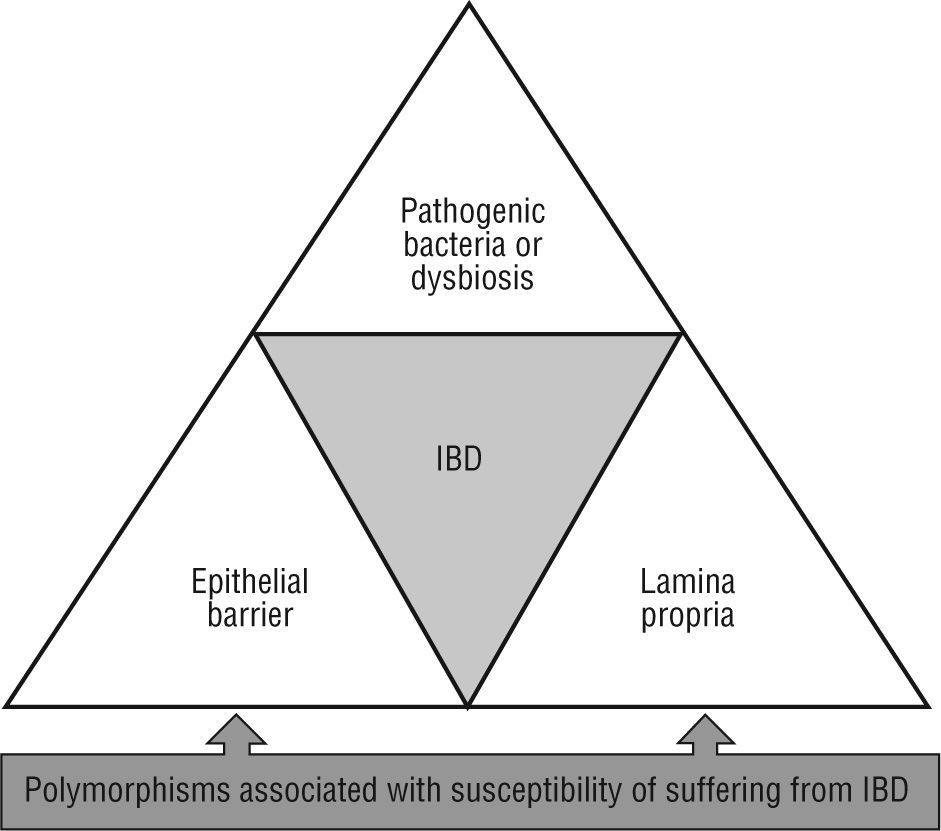
Meanwhile, other essential homeostatic functions of the intestinal epithelium may be seriously compromised by alterations in susceptible genes for suffer IBD such as ATG16L1, OCTN1, OCTN2, MDR1, MUC19, PTGER421,42. Therefore, numerous studies link IBD with the intestinal epithelium damage, which accumulate a set of abnormalities which compromise the isolation of the lamina propria from the external (luminal) milieu (fig. 2).
The influence of TLR-2 mediated actions on the intestinal epithelium
Most of the putative IBD predisposing genetic polymorphisms can alter the correct functioning of both innate and acquired immunity. The family of TLRs is one of the most ancestral systems for pathogen recognition33. TLRs are type 1 transmembrane glycoprotein receptors. The extracellular domain contains consecutive leucine rich repeat motifs involved in ligand binding. The intracellular tail contains a highly conserved region, called Toll-interleukin-1 receptor domain, which mediates interactions between TLRs and downstream signalling molecules (fig. 1). TLRs and other pattern-recognition receptors as NOD-like proteins are an essential element for influencing the innate and adaptive response involved in microbial control. In spite of their presence in a large variety of cells, in dendritic and epithelial cells TLRs play a pivotal role in the homeostasis maintenance. It has been reported in vivo that dendritic cells could increase TLR-activated pro-inflammatory response if they had the NOD2/CARD15 mutated44. Furthermore, polymorphic variants of TLR-4 have been related to the perpetuation of the inflammatory process as well as with a decrease in the repair of intestinal epithelium45-47. However, actions mediated by TLR-2 possess the ability of reinforcing the tight-junctions in epithelial cells (survival and proliferation signals), and inducing tolerogenic responses through dendritic cells (polarizing the immune response towards a T regulator phenotype). TLR-2 forms dimers with TLR-1 and TLR-6 in order to recognise membrane lipoproteins (synthetic or otherwise) of the gram positive bacteria, such as Pam3CSK4 and lipoteicoic acid (LTA), respectively (table 1). TLR-2 uses the adaptor myeloid differentiation primary response gene 88 (MyD88) and toll-IL-1 receptor domain containing adaptor protein (TIRAP)/Mal cytoplasmatic molecules to transmit signals through the IL-1 receptor-associated kinase (IRAK)/ TNF receptor-associated factor 6 (TRAF6)/IKK axis and activates NF-kB-mediated gene expression (fig. 1)43,48.
In IL-10 deficient mice, TLR-2 and MyD88 has been shown to disappear from the intestinal epithelium adjacent to inflamed areas23. This is possibly a consequence of the high concentration of pro-inflammatory cytokines. The reduction of TLR-2 in the intestinal epithelium has also been detected in human colonic epithelial cells and in intestinal epithelial cell lines in response to Gram-positive commensal bacteria, but co-existing with an elevated expression of Toll-inhibitory protein49. All these data suggest the need to suppress the generation of TLR-2-derived pro-inflammatory signals from the intestinal epithelium. Cario et al. recently demonstrated intestinal epithelial cell-specific NF-ĸB downregulation by Pam3CSK4-activating phosphoinositide 3-kinase pathway 50. According to the previous study, enterocyte TLR-2 hyporesponsiveness has been related to NF-ĸB suppression and with the prevalence of mitogen-activated protein kinase p38 pathway51. In short, these studies suggest that TLR-2 employs some mechanisms which are not shared by other TLRs thereby promoting the homeostasis of the intestinal epithelium.
In spite of the potential pro-inflammatory role of TLR-2 activity, it has been related to the production of anti-microbial and defensive peptides by the intestinal epithelium43,48,52. TLR-2 agonists also increase mucosal integrity through para-cellular intestinal epithelium reinforcing50,51,54. Dextran sulfate sodium (DSS)-induced colitis is aggravated in TLR-2 Knockout mice, due to the goblet cells failure to secrete factor-3 trefoil, among other mechanisms2,17. TLR-2 agonists prevent apoptosis of epithelial cells and stimulate damaged mucosa repair17,55. Between growth factors stimulated by TLR-2/trefoil factor-3 axis, the Epidermal Growth Factor (EGF) stands out. This activates its receptor and transmits signals through different adaptor proteins (Grb2, Shc and Sos), Ras, Raf1 and Mitogen-activated protein kinases (Erk1/2). These are capable of activating nuclear factors which increase proliferation and cellular migration56-58. However, this is not the only way to recover the damaged epithelium, given the existence of other complementary mechanisms for mucosal healing59-63.
In our lab, we have also shown that the protection on development of colitis in IL-10-deficient mice treated with medium chain triglycerides or the early recovery of intestinal TNBS-injuries in probiotic-treated mice, are partly mediated by TLR-2/MyD88 64,65. Moreover, the transcriptome (CodeLink™ Mouse Whole Genome Arrays, BIONOVA, Spain) of DSS-treated mice incubated with Pam3CSK4 (via TLR-1/TLR-2) or LTA (via TLR-2/TLR-6) shows a high activation of intestinal epithelium regeneration through Epidermal Growth Factor receptor activation and p53/Kras pathway (Pedrosa E, Lorén V & Mañé J; unpublished data). Additionally, these data coincides with the significant reduction of epithelial damage in DSS-induced colitis mice treated with TLR-2 agonists compared to those that didn't receive them. On the other hand, Pedrosa E et al. showed the key role of the TLR-2/MyD88 axis in the maintenance of intestinal homeostasis in IL-10-deficient mice in a recent study23. Because of this, the role of TLR-2 in the intestinal epithelium recovery represents an attractive therapeutic target regulated by mechanisms which have to be elucidated in the future.
Tolerogenetic actions of dendritic cells on the intestinal mucosaThere are sub-epithelial-derived beneficial actions on intestinal epithelium which come from active myeloid dendritic cells2,9. Our research team has been able to detect a significant increase in TLR-2-stimulated gene expression that mimic dendritic cells activation (chemokine (C-C motif) ligand (CCL) 5, CCL8, chemokine (C-X-C motif) ligand 9, lysyl oxidase, allograft inflammatory factor 1, arginase, resistin like gamma, pentraxin 3, retinol dehydrogenase, etc.) in intestinal inflammatory conditions (Pedrosa E, Lorén V & Mañé J; unpublished data). In an excellent recently published study, IL-22 —a member of the IL-10 family— produced by sub-epithelial dendritic cells (CD11c+), is proven to stimulate the epithelium healing by the signal-transducer and activator of transcription protein (STAT)-3 activation in colitic mice66. However, we have much to learn about the contribution of intestinal dendritic cells activated by TLRs in injured intestinal epithelium. In the bowel, dendritic cells are on continuous alert testing luminal antigens in contact with the intestinal mucus. In order to carry out this function, the dendritic cells establish bidirectional cooperation with the intestinal epithelium, regulating the antigenic presentation and producing a multitude of soluble mediators67-69. As mentioned before, dendritic cell-derived IL-22 influence epithelial cell restoration66, but on other way, factors produced by epithelial cells target on dendritic cells to promote immune-tolerance. Timus stromal lymphopoietin is produced by the epithelial cells and promotes the differentiation of dendritic cells to achieve the development of CD4+CD25+Foxp3+ cells (regulatory T-cells)69. In agreement, timus stromal lymphopoietin was not detected in the intestinal epithelial cells of nearly 70 % of Crohn's disease patients, suggesting a timus stromal lymphopoietin-mediated gut protection70. Epithelial cells-released prostaglandin (PG) E-2 and transforming growth factor (TGF)-b have been shown to reduce T cells-derived cytokines71,72. During infection, epithelial cells release inflammatory mediators, including IL-8 and CCL20. These initiate protective immunity by homing competent CCR6+ dendritic cells from Peyer's patches to respond quickly to invasive pathogens73.
Two phenotypes of lamina propria dendritic cells with a high tolerogenic capacity in the intestine have been recently described 68. CX3CR1+ dendritic cells are able of extending their dendrites through the epithelial paracellular adherences to recognise luminal antigens, while the CD103+ cells transport antigens to the mesenteric lymph nodes favouring the T regulatory or Th-2 responses68,69. These dendritic cells lineages are phenotypically and functionally differentiated from others that occupy follicular structures in the intestine (CD11b+and/or CD11c+). In any case, dendritic cells-derived TLR-2-stimulated IL-10 and TGF-b (together with the expression or blockade of other cytokines or chemokines) polarize inflammatory response towards a regulatory phenotype68-75.
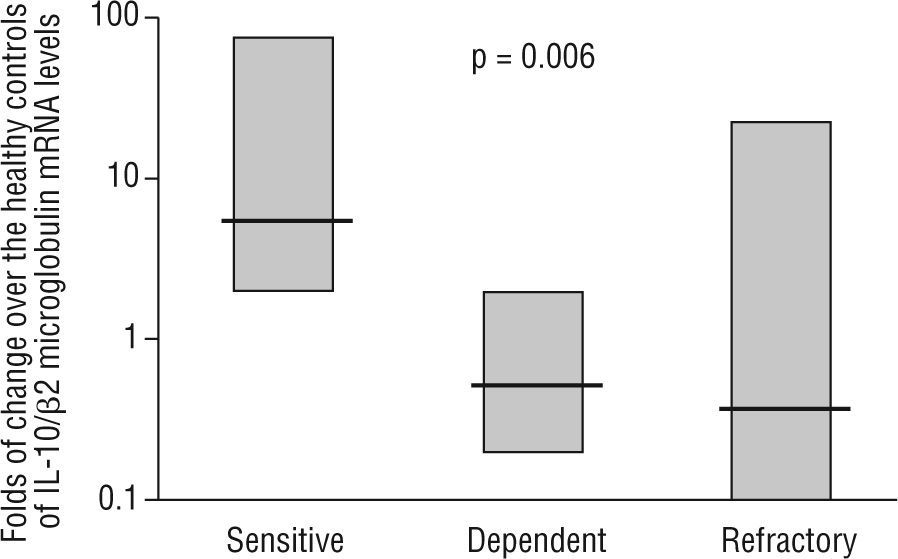
Intestinal epithelial barrier integrity mediated by IL-10Several studies have begun to elucidate the molecular bases through which intestinal dendritic cells are able to drive T cells towards a tolerogenic response. Dendritic cell response is dependent on the IL-10 production to raise CD4+CD25+Foxp3+ cells in the lamina propria74,75. Immune-regulatory activity is dependent on the production of IL-27 and IL-10, as is the case of differentiated dendritic cells via galectine-174. In other cases, TLR-2-activation induces the metabolism of retinoic acid and increases the production of IL-1075. However, recent studies have characterised mechanisms through which dendritic cells would generate a T cell-independent tolerogenic response based on the TLR/interferon (IFN)-1 axis, and its influence on neutrophils and monocytes/macrophages 67. On the other hand, the previous studies also shed light on the extraordinary plasticity of dendritic cells to adapt their response according to their degree of maturity, their location and their inflammatory phenotype2,67,68,74,75. In 1997, Madsen et al. showed how IL-10 protected T84 epithelial cells monolayers integrity against inflammatory mediators such as IFN-g74. It has also been shown the epithelial cells ability to CD1d apical receptor-triggered IL-10 autocrine production77. Recently, it has been reported that the IL-10 or TGF-b neutralization in human colic explants blocks the immunosuppressor gene BCL3 (by producing IL-10) and increases the production of IFN-g, which is harmful for the epithelium78. These studies, together with the increased permeability observed in IL-10 (−/−) mice as prelude of their spontaneous enteropathy79, suggest a protective role of IL-10 on the intestinal epithelium. Additionally, our lab provides new data on the pathogenic IL-10 ablation and key role of TLR-2-mediated intestinal epithelial barrier protection in IL-10 (−/−) mice23. In a recent study, in a series of moderate to severe Crohn's disease patients, we observed that IL-10 mRNA expression in the inflamed mucosa was a predictor of the response to corticosteroid therapy, with an 89 % sensitivity, 67 % specificity and with negative and positive predictive values of 61 % and 91 %, respectively (fig. 3) 24. Therefore, whole evidence suggests a pivotal role of IL-10 on epithelium protection and regeneration. Hence, this would lower the antigenic load reached the lamina propria, and, consequently, diminishing inflammation.
, steroid-dependant (n=7) and steroid-refractory (n=8) Crohn")
mRNA levels of IL-10 in intestinal biopsies before treatment in steroid-responding (n=9), steroid-dependant (n=7) and steroid-refractory (n=8) Crohn's disease patients. Values expressed as median (interquartile range) in logarithmic scale; Kruskal-Wallis test. (Date from reference 24).
IL-10 receptor 1 and 2 triggered Janus kinases downstream, which activate STATs family in epithelial cells 80. Several studies have related STAT-3 phosphorilation to an increase in the restoration of the damaged intestinal epithelium. It has been shown that STAT-3 activity can be induced by the IL-10 family66. Other studies show that IL-10 deficiency inactivates anti-inflammatory mechanisms associated to TGF-β 66,78,81. In this way, a considerable amount of phosphorilated STAT3 has been detected in the intestinal epithelium of IBD patients and in colitis DSS-induced mice80. On the other hand, the allelic variant S138G of IL-10 receptor 1 significantly reduces the phosphorilation of STAT1 and STAT382. These studies also show that IL-10 can also be produced by intestinal epithelium cells suggesting autocrine regulation.
ConclusionsSeveral studies have associated epithelial barrier breakdown with IBD. Within the complex mechanisms of intestinal homeostasis, TLR-2-derived actions are fundamental as a bridge between the recognition of intestinal bacteria and epithelial-activated signalling cell survival. The TLR-2/IL-10 axis in sub-epithelial dendritic cells increases tolerogenic lamina propria lymphocytes and influences mucosal healing through STATs phosphorilation. In spite of the large number of molecular and cellular mechanisms responsible for the maintenance of intestinal mucosa integrity, TLR-2 and IL-10 activation appear pivotal in recovering and reinforcing the epithelium in IBD patients. Consequently, this leads to a decreased number of inflammatory cells in the lamina propria due to decrease of luminal antigens reaching it, thereby facilitating therapeutic efficiency.
Conflict of interestsThe author declares no financial conflict of interests.
I would like to thank Elisabet Pedrosa, PhD and Violeta Lorén, BSc from our Lab, for providing data of their current investigations, as well as Eduard Cabré, MD, PhD for his help in the revision of the manuscript, and the Centro de Investigación Biomédica en Redenfermedades hepato-digestivas (CIBERehd) of the Ministry of Science and Innovation of Spain for the financial support.