






INTRODUCCION
La información sobre la enfermedad de Wilson (EW) proviene de muchas opiniones de expertos basadas en la experiencia, aunque en pocas series de pacientes, con una muestra pequeña, de edad, clínica de comienzo y tratamientos administrados heterogéneos. Actualmente, se lleva a cabo un estudio europeo (EUROWILSON, www.eurowilson.org) para el registro de todos los pacientes de nuevo diagnóstico, dirigido a conocer la incidencia de la enfermedad, las características clínicas y analíticas, y la evolución con el tratamiento disponible. Es un primer paso para conocer en un futuro la influencia de cada mutación, otros genes que puedan modificar la expresión de la enfermedad, y diseñar ensayos de tratamiento. Cada médico que diagnostica un paciente nuevo está invitado a participar (database@eurowilson.org). La información del paciente será introducida en una base de datos a través de los coordinadores de cada país.
La EW es un diagnóstico posible en individuos con hepatopatía y/o trastornos neuropsiquiátricos, principalmente entre 6 y 40 años de edad, aunque con casos aislados de edad muy joven o ancianos. Aunque no está explicado el motivo del muy diferente período en que este trastorno congénito determina la aparición de síntomas clínicos, lo más importante es partir de la idea principal de que es tratable, que el beneficio para el paciente es mucho mayor si se detecta en estados presintomáticos, y que puede y suele perderse la reversibilidad con el tratamiento en estados de hepatopatía complicada, o quedarán secuelas neurológicas si las manifestaciones son muy pronunciadas. Por tanto, debe realizarse siempre un cribado de la EW en cualquier individuo con antecedente familiar o que presente cualquier grado de alteración funcional y hepática, trastorno de comportamiento o del movimiento.
FISIOPATOLOGIA
La base de la EW es el impedimento en la excreción biliar del exceso de cobre.
El cobre es un elemento traza necesario para la estructura y la función de algunas enzimas, pero a la vez es tóxico por su facilidad para intervenir en reacciones de oxidación. La homeostasis del cobre se realiza modulando su eliminación en la bilis1. Hay sistemas de regulación de su absorción intestinal, que disminuye en el caso de depósitos aumentados, pero esos sistemas son insuficientes para evitar la acumulación excesiva de cobre si fracasa la eliminación biliar.
La absorción intestinal del cobre alimentario se realiza con la entrada en el enterocito (transportador CTR1) y el paso a la sangre mediante el transportador ATP7A. Se absorbe un 10-50% del cobre alimentario. En la sangre circula unido a la albúmina y los aminoácidos. En todas las células del organismo, con excepción del hígado, la eliminación del cobre en exceso tiene lugar por transportadores ATP7A hacia la circulación. En el hígado, el sistema es diferente; la entrada mediante CTR1 es seguida de un transporte del cobre mediante chaperonas hacia las enzimas que lo precisan. Un transportador, ATP7B, está localizado en la red trans-Golgi y su función es transportar cobre a través de esa membrana subcelular desde el citoplasma al interior de las cisternas. Ese cobre es cedido a la apoceruloplasmina, y ésta será secretada como holoceruloplasmina a la circulación. La ceruloplasmina tiene una función de movilización de los depósitos de hierro, el cobre que forma parte de esa proteína reducirá el hierro de la ferritina (Fe**) que pasa a Fe+++ para la transferrina. El cobre del citoplasma hepatocitario no utilizado queda fijado por metalotioneína y glutatión para evitar su acción oxidante. El cobre excesivo se elimina por excreción biliar, en su mayor parte contenido en endosomas que se vierten al polo canalicular. La proteína ATP7B es esencial para el transporte del cobre excesivo al interior de los endosomas. Otra proteína (murr1) es necesaria para el movimiento de vesículas y excreción en la membrana canalicular1.
La EW está causada por una mutación en el cromosoma 13, en los 2 alelos del gen ATP7B, que determina las alteraciones en la función doble de la proteína ATP7B (cesión de cobre a ceruloplasmina y eliminación biliar del exceso de cobre). Las mutaciones en el gen ATP7B pueden truncar la síntesis de la proteína, o alterar sitios clave para su función, su configuración terciaria, o su localización dentro del hepatocito. Hasta la fecha, se han descrito más de 200 mutaciones de diverso tipo (deleciones, inserciones, missense, nonsense, splice site)2. También se han identificado en la región 5' no codificante. Hay una elevada tasa de individuos en los que no se identifican alteraciones en los 21 exones, por lo que se sospecha que otras mutaciones hasta ahora no identificadas se localicen en las regiones reguladoras del gen.
El fracaso de la función de ATP7B determina 2 consecuencias. La principal es la retención de cobre en exceso dentro del hepatocito. Se origina un daño oxidativo en las organelas subcelulares, más precoz en las mitocondrias, que sufren trastornos en la cadena respiratoria y en la fosforilización oxidativa por el exceso de radicales libres. Además, la síntesis de ceruloplasmina completa está disminuida y la proteína defectuosa en el cobre se detecta de forma incompleta por las técnicas habituales (nefelometría) y tiene una vida media acortada, por lo que el valor sérico es bajo.
Los mecanismos para compensar el exceso de cobre son el descenso de su absorción intestinal y el aumento de la fijación del metal en el citoplasma de manera no tóxica, incrementando la tasa de saturación en cobre de la metalotioneína.
El daño en el hepatocito causado por el cobre, en ocasiones disparado por otros motivos de lesión concomitantes, permite la salida de éste a la circulación. Aumenta en el suero la fracción de cobre libre y su llegada a otros tejidos, que son dañados. El tejido cerebral, y dentro de él los núcleos de la base lenticular y putamen, son los más sensibles a la toxicidad oxidativa del cobre. El depósito de cobre en la membrana de Descemet origina una imagen peculiar, el anillo de Kayser-Fleischer, normalmente sólo visible mediante exploración con lámpara de hendidura y en los pacientes que ya tienen lesiones en los núcleos de la base. Además del sistema nervioso, con cifras elevadas de cobre libre sérico, hay manifestaciones de lesión en las células tubulares renales, dolor articular, osteoporosis y hemólisis.
No se conocen bien los factores que influyen en la edad y los síntomas de presentación de los pacientes. Pueden depender, en parte, de la cantidad de cobre en la dieta o radicar en diferencias genéticas de la capacidad para inducir metalotioneína o enzimas protectoras de oxidación. La enfermedad es de penetrancia casi completa, los individuos con dos mutaciones del gen ATP7B tienen casi un 100% de riesgo de desarrollar enfermedad.
CLASIFICACION DE LA ENFERMEDAD DE WILSON SEGUN LOS SINTOMAS
Los signos de enfermedad siguen una secuencia dependiente del depósito de cobre; por tanto, comienzan por hepatopatía, siguen con la aparición de anillo de Kayser-Fleischer y trastornos neuropsiquiátricos. Los síntomas de consulta son principalmente hepáticos o neurológicos, ya que en muchos pacientes con afección cerebral la participación hepática no ha derivado en complicaciones, aunque histológica o funcionalmente sea grave.
En 2002 tuvo lugar una reunión de expertos dirigida a clasificar los síntomas de pacientes con EW, buscando una manera de progresar en la investigación de la relación de determinadas mutaciones de ATP7B con el fenotipo clínico, la precocidad y la gravedad de la enfermedad. La clasificación fenotípica queda reflejada en la tabla I3.

DIAGNOSTICO DE LA ENFERMEDAD DE WILSON
El diagnóstico de EW se basa en la combinación de rasgos clínicos, pruebas de laboratorio y análisis mutacional. Un panel de expertos elaboró un sistema de puntuación que tiene interés para los pacientes que consultan por enfermedad hepática, cuyas pruebas diagnósticas habituales no son concluyentes (tabla II)3.

Las pruebas básicas para el cribado de la EW son la determinación de ceruloplasmina, de cobre total en suero y cobre en orina de 24 h4. Un paciente típico presenta una ceruloplasmina baja (< 20 mg/dl) y un descenso del cobre total en suero, dependiente de la cifra baja de ceruloplasmina, ya que esa proteína contiene más del 95% del suero determinado en suero. El cobre en orina está aumentado en muchos pacientes, pero depende del aumento de cobre libre en suero. No hay habitualmente posibilidad de determinar el cobre libre en suero, sólo de estimarlo (cobre libre = cobre total sérico [ceruloplasmina 3]).
El diagnóstico es complicado en circunstancias que artefactan la cifra de ceruloplasmina, que es un reactante de fase aguda y puede estar falsamente aumentado en condiciones de inflamación, colestasis e ingesta de anovulatorios. En un paciente con hepatopatía ictérica la cifra de ceruloplasmina puede ser normal, por lo que cobra especial importancia la determinación de cobre en orina de 24 h, que habitualmente está muy elevada.
El diagnóstico también es complejo en los pacientes absolutamente asintomáticos (p. ej., en hermanos de un niño afectado). Las determinaciones para descartar una EW no pueden realizarse en niños menores de un año, pues dentro del metabolismo de cobre normal para esa edad hay cifras bajas de ceruloplasmina en suero hasta los 6 meses, y cifras más altas de lo normal después; asimismo, la medición de cobre hepático indica depósitos aumentados en los lactantes pequeños sanos; ese cobre está fijado por metalotioneína y corresponde a un proceso normal de acumulación fetal para hacer frente a la vida posnatal inicial, en la que la leche aporta cobre en muy escasa cantidad5. En los niños mayores de un año, y hasta la edad joven, el estudio para descartar una EW puede mostrar todos los parámetros normales, pero cambiar evolutivamente hacia un descenso de ceruloplasmina. Por tanto, es recomendable un chequeo periódico es los sujetos jóvenes aparentemente normales que son familiares de un paciente con EW. Además, un 10% de los portadores heterocigotos pueden tener cifras bajas de ceruloplasmina.
La biopsia hepática puede aportar datos compatibles con la EW, pero los cambios histológicos no son específicos. El cobre hepático aumentado puede ser apreciado por técnicas tintoriales para cobre (rodanina, rubeánico) o proteína asociada a cobre (orceína), pero solamente son positivas en una minoría de niños con EW, casi siempre restringido a los que presentan una lesión muy avanzada.
La prueba más fiable para la confirmación de EW es la cuantificación de cobre en hígado. El tejido se obtiene mediante biopsia con aguja, pero es necesario un mínimo de 1 cm de longitud. La aguja y el contenedor deben estar libres de cobre. Mediante espectrofotometría, el cobre normal en el hígado es inferior a 50 µg por gramo de tejido seco. En la EW hay más de 250 µg/g, y en los pacientes heterocigotos pueden encontrarse cifras entre 50 y 250 µg/g. A pesar de ser el patrón estándar del diagnóstico, hay también posibilidades de falsos positivos y negativos. En pacientes colestáticos de larga evolución por hepatopatías diferentes de EW se produce también aumento del cobre en el tejido, debido a la dificultad de eliminación biliar, aunque ese cobre está procesado por ATP7B e incluido en vesículas que atenúan su toxicidad. Por otra parte, en pacientes con EW y hepatopatía avanzada puede haber una fibrosis extensa, y esas zonas incluidas en la biopsia motivan una cuantificación de cobre menor de la que hay en el tejido; además, la distribución de cobre puede ser muy variable entre nódulos cirróticos. Por motivos no explicados, la determinación del cobre en el teji do ha sido normal en casos aislados de niños que cumplían otros criterios suficientes para el diagnóstico de EW y no presentaban cirrosis. Un reciente estudio realizado en pacientes con EW, comparados con pacientes con hepatopatías no colestáticas (hepatitis C y esteatohepatitis no alcohólica), mostró que el cobre en el tejido era > 250 µg/g en el 83,3% de los pacientes con EW y en un 1,4% de los afectados por otra hepatopatía; mientras que los valores de 50-250 µg/g se encontraron en el 9,1% de los pacientes sin EW6. Con estos datos se refuerza la idea de que el diagnóstico de EW precisa la combinación de diversas pruebas y datos clínicos.
El análisis mutacional comienza a contribuir en el diagnóstico confirmatorio de los pacientes y en el cribado de los hermanos. El trabajo realizado desde la identificación del gen en 1993 permite que en ciertas zonas geográficas concretas pueda hacerse una búsqueda relativamente rápida de las mutaciones más prevalentes. En Europa central y del este la mutación H1069Q se encuentra en el 50-80% de los pacientes3. En España la mutación más frecuente es Met645Arg, detectada en el 55% de los casos, pero en un estudio sobre 64 individuos de 40 familias se observaron 21 mutaciones diferentes. Sin embargo, un análisis de las mutaciones hasta ahora descritas que resulte completamente negativo no excluye el diagnóstico, ya que el conocimiento de las mutaciones no es completo en la actualidad. En el estudio de pacientes españoles lograron caracterizarse el 74% de los alelos de la enfermedad7.
ENFERMEDAD DE WILSON EN LOS NIÑOS
En la literatura médica clásica sobre la EW, los síntomas de los pacientes son de hepatopatía en el 40% de los casos, de alteración neuropsiquiátrica en el 50%, y de otros órganos (anemia, tubulopatía) en el 10%. Son individuos característicamente jóvenes, entre 10 y 40 años de edad (rango, 3-60). En el caso de los niños menores de 10 años, el 83% consultaba por hepatopatía, mientras que en los adolescentes había un 50% de casos hepáticos y un 50% neurológicos; en mayores de 18 años la enfermedad neurológica era el motivo de consulta en el 74% de los pacientes8.
En las últimas décadas, el diagnóstico de EW es más precoz y el porcentaje de niños detectados antes de que aparezcan los síntomas es cada vez mayor. Es una práctica habitual la búsqueda de EW en niños que presentan una elevación de las aminotransferasas sin otros signos o datos sobre la enfermedad. En un estudio italiano9, de 425 niños consecutivos atendidos por presentar una elevación aislada de aminotransferasas, la EW fue diagnosticada en 18 casos.
Las series de pacientes pediátricos menores de 18 años indican en España e Italia que el diagnóstico se realiza en la etapa presintomática en un 75-80% de los casos. En el 20-25% de los casos hay síntomas, un 75-92% son de hepatopatía y un 8-25% de alteración neurológica10,11.
Enfermedad presintomática
El hallazgo incidental de alteraciones leves en la función hepática es el patrón más frecuente. Probablemente, todavía está poco extendido el cribado de EW en niños que presentan cambios neurológicos leves. Debería indagarse la enfermedad también en los niños que presentan cambios en su comportamiento, un deterioro del rendimiento escolar o de la coordinación en actividades manuales finas.
A pesar de que la acumulación de cobre se debe a un trastorno congénito, algunos casos mantienen enteramente normales los parámetros de función hepática, al menos hasta los 18 años, sin evidencia de lesión en la biopsia. Los primeros signos de lesión son evidentes mediante microscopia electrónica. Hay alteraciones en las mitocondrias, que aparecen hinchadas, con separación de la membrana interna, gránulos en su interior y crestas gruesas. En la microscopia óptica el primer dato es la glucogenización nuclear.
En pacientes pediátricos con elevación de las transaminasas y ausencia de datos clínicos, la biopsia hepática detecta lesiones con infiltrado inflamatorio crónico portal y citólisis lobulillar; además, un 75% de los casos tienen esteatosis y fibrosis10. A pesar de no presentar síntomas, la fibrosis muestra un estadio avanzado en el 15% de los casos.
En los pacientes con normalidad funcional es difícil confirmar la presencia de EW, ya que la mitad no tiene ninguna alteración en las pruebas habituales, o presenta cifras ocasionalmente límites de ceruloplasmina; por ello, es necesario basar el diagnóstico en la cuantificación de cobre en el tejido. Por el contrario, los pacientes asintomáticos con disfunción hepática son los más fáciles de diagnosticar, mediante la determinación de ceruloplasmina (baja en el 96% de los casos), cobre sérico (bajo en el 81% de los casos), cobre libre deducido (alto [> 10 µg/dl] en el 69% de los casos) y cobre urinario (en el 68% elevado [> 100 µg/día]). Las cifras de cobre en el tejido oscilan en nuestra experiencia entre 225 y 1.870 µg por gramo de tejido seco.
En la experiencia del King's College, los pacientes asintomáticos con EW que fueron detectados en estudio familiar presentaban una elevación de las transaminasas en el 65% de los casos, un 35% de gamma glutamiltranspeptidasa (GGT) elevada, y parámetros normales de síntesis hepatocelular. La cifra de ceruloplasmina era baja en el 88,2%, la cupruria elevada en el 70,6%, y el cobre hepático también elevado en el 87,5%12. La prueba de penicilamina, que consiste en la medición de cobre en orina de 24 h, recogida desde la administración de 500 mg de penicilamina con segunda dosis de 500 mg 12 h después13, fue positiva (> 25 µmol/día) en un 55,6% de esos niños.
Hepatopatía sintomática
Los síntomas de hepatopatía se correlacionan con una enfermedad hepática histológicamente avanzada, con cirrosis en todos los casos, asociada con una notabale inflamación portal en la mayoría de ellos, mientras que, a diferencia de los pacientes menos evolucionados, es raro observar esteatosis. Los pacientes pueden consultar por la presencia de ictericia, ascitis o sangrado. Analíticamente, tienen parámetros de insuficiencia hepatocelular y cifras elevadas de aminotransferasas. Puede haber anemia hemolítica, y signos de tubulopatía.
En ocasiones, los pacientes parecen presentar un fallo hepático agudo. Esa forma de presentación corresponde a un proceso de necrosis lobulillar que incide sobre un fondo de cirrosis establecida. Hay síntomas graves de inicio brusco en pacientes que no aparentaban tener una hepatopatía previa, con insuficiencia hepatocelular y hemólisis. Este cuadro se ha caracterizado analíticamente por una disociación de las cifras de fosfatasa alcalina, bajas, en contraste con la ictericia. Berman et al14 detectaron que la cifra de fosfatasa alcalina era, como media, de 49 U/l y la de bilirrubina de 43 mg/dl, unos valores respectivamente menores y mayores que los presentados en pacientes adultos con otras etiologías del fallo hepático. La cifra de transaminasas también fue significativamente menor, con una alanina transferasa (ALT) media de 22 y aspartato-aminotransferasa (AST) de 122 U/l. Aunque otros casos recogidos en la bibliografía presentaban cifras más elevadas, los valores de AST siempre eran superiores a los de ALT. Característicamente, se observan valores de cobre sérico normal o alto a expensas del aumento de cobre libre. La presentación «fulminante» ha conllevado el fallecimiento de todos los casos tratados médicamente, con tratamiento quelante de cobre y todas las medidas de soporte avanzado. La identificación de EW con esta forma de inicio indica la realización de un trasplante hepático urgente4.
En una serie amplia12 de niños ingleses que presentaban síntomas de enfermedad hepática al diagnóstico, la edad media fue de 12 años (el más joven tenía 6 años). El 47% de los 57 casos comunicados manifestaban un fallo hepático agudo, con signos de encefalopatía en la mitad de ellos. En los niños, el concepto reciente de «fallo hepático agudo» es diferente del de los adultos, y no es imprescindible la coexistencia de encefalopatía, debido a que esa complicación es difícil de reconocer en niños pequeños o aparece muy tardía en el curso de la enfermedad. Del total de 57 niños, 41 presentaron ictericia, 19 ascitis y uno hemorragia. El 30% tuvo anemia hemolítica Coombs negativa. Las pruebas de diagnóstico dieron como resultado una ceruloplasmina baja en el 77% de los niños, un cobre urinario elevado en el 95% de los casos, y un aumento de cobre en el tejido hepático en el 82% de los analizados. En 11 niños se detectaban autoanticuerpos circulantes del tipo antinucleares y/o antimúsculo liso principalmente. La positividad de los autoanticuerpos es inespecífica, pero podría dar lugar al diagnóstico erróneo de hepatitis autoinmune si no se realizaran pruebas adicionales para identificar la EW.
Manifestaciones neuropsiquiátricas
Las manifestaciones neurológicas de EW son muy multiformes. Los signos clásicos consisten en manifestaciones de disfunción extrapiramidal o cerebelosa. Las anomalías motoras son notables, y no hay anomalías sensitivas. Los síntomas observados pueden ser temblor de reposo e intención, dificultad para articular las palabras, salivación excesiva, rigidez, distonía, disfagia, ataxia y movimientos coreiformes. Suelen estar precedidos por cambios de carácter o de rendimiento intelectual unos años antes, que pueden inducir una primera consulta al psicólogo o psiquiatra5.
Tradicionalmente se consideraba excluida la EW en pacientes con síntomas neuropsiquiátricos con una edad mayor de 30 años al comienzo, sin anillo de Kayser Fleisher. Hay casos aislados en la literatura médica que demuestran que éstas no son condiciones excluyentes, aunque la mayoría de los pacientes acude por primera vez a consulta entre los 13 y los 20 años. El anillo de Kayser se encuentra al menos en un 90-100% de los casos. Es necesaria la observación con lámpara de hendidura por parte de un oftalmólogo experto. En otros pacientes de ojos claros puede observarse a simple vista un reborde marrón en la periferia de la córnea. Todos tienen hepatopatía evidente por alteraciones analíticas, clínicas y/o histológicas. En algunas series todos los pacientes con síntomas neurológicos presentaban cirrosis subclínica.
PRONOSTICO DE LOS NIÑOS CON ENFERMEDAD DE WILSON
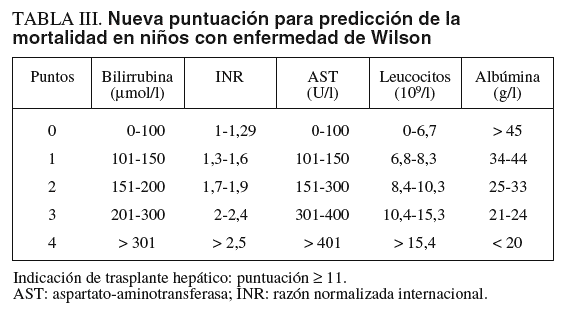
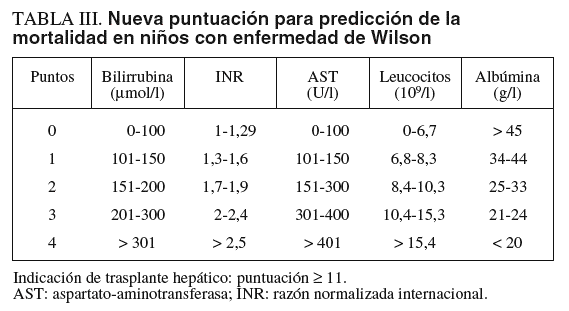
El tratamiento médico con penicilamina, trientina o sales de cinc permite obtener una normalidad funcional del hígado en un plazo de 1-2 años en niños con EW detectada en etapas presintomáticas. En niños con hepatopatía avanzada sintomática la tasa de respuesta al tratamiento es del 50%10,12. La identificación de los niños con posibilidad nula o baja de responder a la terapia es fundamental para indicar la realización de trasplante hepático. Recientemente, se han revisado las características clínicas y analíticas que se asocian a una escasa respuesta al tratamiento. Una nueva puntuación sustituye a la previo de Nazer et al15, basada en la cifra de bilirrubina, AST y razón normalizada internacional (INR) que tenía para una puntuación > 7, una sensibilidad del 87% y una especificidad del 90%, con un valor predictivo positivo del 72% de mortalidad a pesar del tratamiento médico. En la revisión de la puntuación se analizaron 32 niños con hepatopatía sintomática que sobrevivieron con tratamiento y 15 niños que fallecieron. Mediante el cálculo con curvas ROC y análisis multivariado, los autores concluyeron que con la combinación de AST, albúmina, bilirrubina, INR y recuento de leucocitos se realizaba una mejor predicción de la mortalidad (tabla III)12. Según el nuevo sistema de puntuación, 11 o más puntos tenían una sensibilidad del 93%, una especificidad del 97%, un valor predictivo positivo del 92% y un valor predictivo negativo del 97%. Este nuevo «Wilson Index» fue aplicado prospectivamente a 14 casos pediátricos sintomáticos, entre 6 y 16 años de edad. Ninguno de los que respondieron al tratamiento tuvo una puntuación > 6; de los 5 casos con puntuación > 11, uno respondió al tratamiento y 4 fueron trasplantados. En el estudio prospectivo la nueva puntuación tuvo una sensibilidad del 72% y una especificidad del 91%.
El trasplante hepático es necesario en un 8-18% del total de los niños con EW, y en un 50-60% de los pacientes con síntomas de hepatopatía al diagnóstico; el trasplante cura la hepatopatía y la anomalía de ATP7B, por lo que no es necesario el tratamiento quelante posteriormente16. En niños trasplantados de donante vivo se ha descrito un fenotipo de portador correspondiente al padre donante, con cobre hepático aumentado, pero inferior a 250 µg/g17.
El trasplante ofrece una normalización completa del metabolismo del cobre, pero ello no permite una mejoría de las secuelas neurológicas en los casos con manifestaciones neuropsiquiátricas, por lo que no está indicada su realización.
TRATAMIENTO MÉDICO DE NIÑOS CON ENFERMEDAD DE WILSON
Los fármacos disponibles son penicilamina, trientina y sales de cinc. El tratamiento tiene que prolongarse toda la vida. Explicar a la familia y al paciente la trascendencia de la medicación y un seguimiento frecuente reforzando ese hecho en cada visita es fundamental a todas las edades, sobre todo en los jóvenes. El valor de una reducción del cobre en la dieta en pacientes que reciben tratamiento es controvertido. Parece razonable excluir los alimentos más ricos en cobre, como el chocolate, el hígado de cualquier animal y los frutos secos, pero los pacientes bien controlados podrían consumirlos de forma limitada.
Penicilamina
La penicilamina es un metabolito de penicilina (ß-ß-dimetilcisteína). Es el fármaco más potente en el tratamiento de la EW y el mejor conocido de los disponibles. La penicilamina forma un quelato soluble con el cobre que se elimina por la orina18. La cupruria es mayor en las primeras semanas tras el comienzo de la terapia porque hay más cantidad de cobre libre susceptible de ser quelado, y continúa en el seguimiento de los pacientes, en torno a 200-500 µg/día4. No hay casos de deficiencia en cobre debida a este tratamiento quelante si el paciente padece EW. Actualmente se considera que, además de producir una eliminación de cobre en orina, el efecto principal de la penicilamina es la inducción de metalotioneína en el hígado, lo que permite fijar cobre y disminuir su efecto tóxico. Ello explica que las biopsias sucesivas bajo tratamiento con penicilamina muestren una cuantificación de cobre con poco o ningún cambio respecto a la obtenida antes de tratar5, aunque la lesión histológica mejora, y que los individuos tratados durante años puedan presentar cuadros de deterioro hepático brusco poco después de suspender la penicilamina (entre 2 meses y 3 años). La metalotioneína tiene una vida media corta, inferior a 12 h, y mantener su producción inducida resulta prioritario durante toda la vida del paciente.
La penicilamina se emplea en niños en dosis de 10 mg/kg/día los primeras 2 semanas para observar una posiacute;mite máximo de 1 g/día. Para favorecer el cumplimiento terapéutico, en pacientes con una función hepática ya normalizada puede espaciarse la administración cada 12 h. La penicilamina puede causar una deficiencia clínica de piridoxina, por lo que habitualmente se asocia piridoxina 25-50 mg/día6.
La eficacia y el cumplimiento del tratamiento se controlan mejor por criterios clínicos. En los niños se obtiene normalidad de todos los parámetros analíticos hepáticos alrededor de un año después de iniciar tratamiento; en los casos con una grave afección, la normalidad de albúmina y la coagulación se obtienen en meses, pero con frecuencia persiste una ligera alteración de las transaminasas. Hay pocos estudios histológicos evolutivos, que indican que la esteatosis desaparece en individuos tratados, así como la inflamación portal. Los adolescentes presentan un riesgo de incumplimiento terapéutico significativo, pero no hay posibilidad fiable de confirmar el incumplimiento mediante pruebas de laboratorio. Las cifras de cupruria muy elevadas pueden corresponder a un mal cumplimiento, si el paciente toma la medicación en los días previos a la revisión médica. La aparición de anillo de Kayser o de síntomas hepáticos o neurológicos en un paciente que no los presentaba previamente son signos de mal cumplimiento terapéutico.
El control de la toxicidad de penicilamina se realiza con el análisis periódico de hemograma, sedimento urinario y exploración física. Algunas manifestaciones de toxicidad, como urticaria o artralgias, pueden ceder con la asociación de esteroides o antihistamínicos durante unos días, suspensión de penicilamina y reintroducción gradual al controlar el efecto adverso. Actualmente, la disponibilidad de otros fármacos permite la sustitución de penicilamina si ésta determina un efecto adverso significativo. Los efectos tóxicos de la penicilamina afectan a un 10-20% de los pacientes, y se observan, en su mayoría, en las primeras 3 semanas de tratamiento. Los efectos precoces más comunes son aparentemente alérgicos: fiebre, exantema maculopapular, linfadenopatía, leucopenia o trombopenia. Otros efectos adversos posibles son muy diversos y tardíos: ageusia, anorexia, náuseas, estomatitis, anemia, trombopenia, leucopenia, alopecia, onicopatía, pénfigo, lupus, elastosis serpiginosa, síndrome nefrótico, artritis reumatoide, Goodpasture, miastenia o fibrosis pulmonar. Un 10-50% de los pacientes que presentan síntomas neuropsiquiátricos debidos a la EW presenta un empeoramiento de esos síntomas al comienzo del tratamiento con penicilamina, y aproximadamente un 25% tiene después secuelas de ese aumento de síntomas que está relacionado con la movilización del cobre en las etapas iniciales de tratamiento.
Trientina
Es un quelante de cobre con una acción menos potente que la D-penicilamina en cuanto a la cantidad de cobre eliminado en orina, pero clínicamente eficaz y considerada una terapia de primera línea. Se ha usado con más frecuencia en pacientes con algún efecto adverso grave causado por la penicilamina. Se utiliza en niños en dosis de 20 mg/kg/día repartida en 3 tomas, separada de las comidas.
Sales de cinc
El mecanismo de acción del cinc es dificultar la absorción intestinal de cobre. Induce metalotioneína en el enterocito, y se fija el cobre en el citoplasma. El complejo del metal con metalotioneína se perderá en las heces junto con las células intestinales descamadas, en 6 días de promedio. Otro efecto del cinc es la inducción de metalotioneína en el tejido hepático.
La acción de las sales de cinc es lenta para producir el comienzo en el bloqueo de absorción. Esa característica limita su uso como único compuesto farmacológico en pacientes con síntomas, en los que se busca la acción más rápida posible.
Actualmente, las sales de cinc se consideran un tratamiento válido, pero restringido al tratamiento de mantenimiento de los pacientes asintomáticos, porque han sido diagnosticados en esa fase o porque han mejorado tras recibir penicilamina o trientina. En esa situación se consideran igual de eficaces que la penicilamina o la trientina para sostener la mejoría del paciente o la normalidad4.
En los pacientes sintomáticos, algunos autores han recomendado la asociación de cinc con trientina. El objetivo es evitar la penicilamina y sus posibles efectos adversos. Es complejo usar 2 fármacos que precisan 3 administraciones diarias separadas de las comidas, y no hay pruebas que confirmen unos resultados mejores que con el empleo aislado de penicilamina o trientina. La asociación de penicilamina y cinc es también posible, buscando la mayor rapidez en el efecto terapéutico.
Brewer et al19 han descrito la experiencia favorable en 34 niños tratados con acetato de cinc, de edad entre 3 y 17 años. De ellos 17 eran presintomáticos, 7 tenían síntomas de hepatopatía y 10 de alteración neurológica. Después de suspender el tratamiento quelante que algunos habían recibido, se administró cinc. Hasta los 5 años, la dosis de cinc fue de 25 mg 2 veces al día, entre los 6 y 15 años de 25 mg 3 veces al día, y de 50 mg 3 veces al día en los mayores de 16 años. La cifra de transaminasas se redujo tras un año de tratamiento y fue sostenida después, sin deterioro en ningún paciente.

