






INTRODUCCIÓN
La protoporfiria eritropoyética (PPE) es una enfermedad genética de la síntesis del hem caracterizada por una producción excesiva de protoporfirina (PP), en la que se puede dañar el hígado como consecuencia de la acumulación de estas porfirinas1. La enfermedad es debida a un déficit parcial de la actividad de la ferroquelatasa, causada por un defecto, deleción y/o mutación, del gen que codifica la síntesis de esta enzima. Se hereda de manera autosómica dominante con un grado variable de penetración, aunque se han descrito casos de herencia autosómica recesiva. La expresión clínica de la enfermedad ocurre cuando la actividad ferroquelatasa se reduce en más del 30%. Esta reducción de la actividad de la enzima, mayor de la esperada en un heterocigoto, se atribuye a una herencia combinada de un alelo mutado con un alelo normal de baja expresión2,3.
La reducción de la actividad ferroquelatasa, que es la enzima de la que depende la incorporación del hierro a la PP para formar el hem, determina una acumulación masiva de PP en eritrocitos, plasma y heces4. El exceso de PP se origina en los eritrocitos inmaduros, normoblastos y reticulocitos de la médula ósea. El hígado es el órgano responsable de la excreción de la PP. La carga diaria de PP que alcanza el hígado está formada por la suma de las que llegan por primera vez al hígado procedentes del plasma y las que ya han experimentado una circulación enterohepática.
Las moléculas de PP son excitadas al absorber la energía lumínica, lo cual determina la formación de radicales libres, que son los responsables de la lesión en los tejidos. Las moléculas de PP alcanzan la piel por difusión a través del plasma. La exposición a la luz excita las porfirinas depositadas en la dermis, así como las porfirinas intraeritrocitarias cuando los eritrocitos atraviesan los vasos capilares de la dermis, que tienen un calibre inferior a su diámetro1.
EXPRESIÓN CLÍNICA
La enfermedad se caracteriza por su inicio en la infancia en forma de fotosensibilidad cutánea de las áreas expuestas a la luz. Los síntomas son prurito, sensación de quemazón y dolor que aparecen casi inmediatamente después de la exposición al sol, persisten varios días y dificultan la actividad personal y el sueño. Esta fotosensibilidad induce cambios cutáneos como edema y eritema en las zonas expuestas, y sólo raramente, formación de ampollas, que es lo propio de la porfiria cutánea tarda. Una cuarta parte de los casos presentan una anemia microcítica e hipocroma moderada5.
La intensidad de los síntomas es muy variable de unos pacientes a otros, y también en un mismo paciente de un día a otro. Igualmente es variable la edad de presentación. Por lo general se manifiesta ya en la primera infancia, pero en algunos casos aparece en la adolescencia o incluso en la edad adulta.
Con la progresión de la enfermedad, si el paciente no evita la exposición solar, aparece un engrosamiento de la piel, que adquiere un aspecto como de un empedrado6.
Por lo común tiene un curso benigno y sólo provoca molestias en primavera y verano, pero algunos pacientes tienen complicaciones hepáticas graves, lo que justifica una vigilancia sistemática de la función del hígado.
DIAGNÓSTICO BIOQUÍMICO
La confirmación diagnóstica de la PPE exige la demostración de una elevación del contenido en la PP libre (no asociada al zinc) de los hematíes y en el plasma, así como en las heces, ya que el exceso de PP en el plasma se elimina mediante la secreción biliar. Aunque parte de la carga de PP eliminada con la bilis se reabsorbe en el íleon, el resto llega a las heces, con lo cual éstas también tienen un elevado contenido de PP. Las concentraciones de las demás porfirinas son normales.
AFECTACIÓN HEPÁTICA
Manifestaciones clínicas
Una cuarta parte de los pacientes con PPE llegan a desarrollar algún tipo de afectación hepática. Cuando es leve, como sucede en el 20% de los casos, se manifiesta por un aumento discreto de las enzimas hepáticas, tanto de las transaminasas como de las enzimas de colestasis. Las formas graves ocurren en un 5% de los pacientes con PPE y se caracterizan por un cuadro rápidamente progresivo de insuficiencia hepatocelular, que conduce en pocas semanas (de 4 a 12) al fallecimiento del paciente, motivo por el cual está indicado proceder a un trasplante hepático7.
El cuadro de insuficiencia hepática se inicia con ictericia y fatiga intensa, a menudo acompañada de dolor en el epigastrio con irradiación a la espalda. En las semanas siguientes se instauran una ascitis y frecuentemente encefalopatía. La fotosensibilidad se hace más intensa. Los exámenes de laboratorio muestran elevación de la bilirrubina, transaminasas y fosfatasas alcalinas, signos de hiperesplenismo y reducción de la tasa de protrombina8. El desencadenante del episodio puede ser una coledocolitiasis, pero otras veces se desarrolla sin ningún factor desencadenante.
La mayoría de los pacientes que presenta esta complicación tiene más de 30 años y se les diagnosticó muchos años antes de enfermedad hepática por la detección de una elevación asintomática de las enzimas hepáticas; sólo se ha observado algún caso aislado en niños. El examen histológico del hígado en estos casos siempre revela la existencia de cirrosis.
Un 10% de los pacientes con PPE presenta colelitiasis, que puede dar lugar a manifestaciones clínicas igual que las litiasis de colesterol o las pigmentarias9. Los cálculos son birrefringentes debido a su contenido en PP.
Patogenia de la lesión hepática
Las manifestaciones de insuficiencia hepática aparecen cuando se alcanza el punto crítico en que el hígado ya no es capaz de excretar el exceso de PP producido por el paciente, debido a una interferencia en la secreción biliar provocada por la misma PP o por otra causa de obstrucción biliar.
Los hepatocitos captan la PP del plasma por difusión y la eliminan por la bilis. No obstante, la PP inhibe el flujo biliar independiente de las sales biliares, posiblemente por su acción sobre la enzima adenosintrifosfato dependiente de sodio y potasio (ATPasa Na-K), enzima que interviene en la secreción biliar induciendo su retención en los hepatocitos. La captación de PP del plasma persiste aunque esté alterada su eliminación por la bilis, de modo que de forma progresiva se acumulan mayores cantidades de PP en los hepatocitos, donde ejercen un efecto tóxico10. Cuando aparece ictericia, que indica una dificultad extrema en la secreción biliar, la acumulación de PP aumenta de un modo muy acentuado, lo que explica el curso rápidamente progresivo de la insuficiencia hepática, y se produce también un reflujo de PP al plasma, con el consiguiente empeoramiento de la fotosensibilidad cutánea, la posible aparición de un trastorno neurológico agudo, que se parece a una neuropatía axonal generalizada11,12, y de hemólisis13. Ésta es debida al elevado contenido en PP de los hematíes, lo que los hace más sensibles al estrés fotooxidativo. La concentración de PP fecal disminuye en paralelo al empeoramiento de la función hepática, que impide su eliminación biliar.
Se desconoce cuál es la razón por la que unos pacientes presentan una lesión hepática avanzada y otros no, aunque puede guardar relación con el tipo de defecto genético en el gen de la ferroquelatasa.
Biopsia hepática
La biopsia hepática es útil para confirmar la existencia de afectación hepática en la PPE, ya que permite comprobar el depósito de PP, que se visualiza en forma de material pigmentario intrahepatocitario y de depósitos de color marrón en la luz de los canalículos biliares y en macrófagos portales, muy parecidos a los trombos biliares, de los que se distinguen en las preparaciones teñidas con hematoxilina-eosina por su color más marrón14 (fig. 1). Macroscópicamente, el depósito de PP confiere al hígado un color negro cuando alcanza la fase de cirrosis.
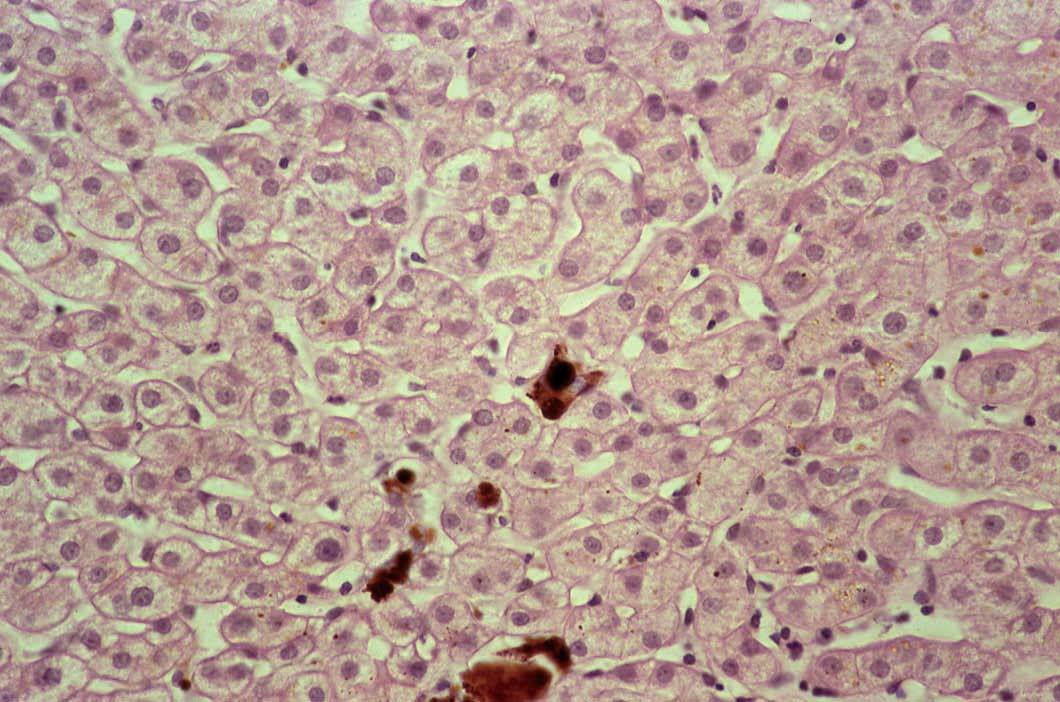
Fig. 1. Depósitos de material de color marrón en la luz dilatada de canalículos biliares, más oscuros que los trombos biliares, que corresponden a acumulaciones de protoporfirinas. (Hematoxilina-eosina, *240.)
El examen con microscopia de luz polarizada demuestra la birrefringencia de la PP. Los depósitos puntiformes aparecen brillantes con la polarización, y los de mayor tamaño, en forma de cruz de Malta, que es una imagen patognomónica de esta enfermedad15 (fig. 2). El examen del cilindro de biopsia mediante lámpara de Wood da una fluorescencia roja, que traduce la presencia de PP.
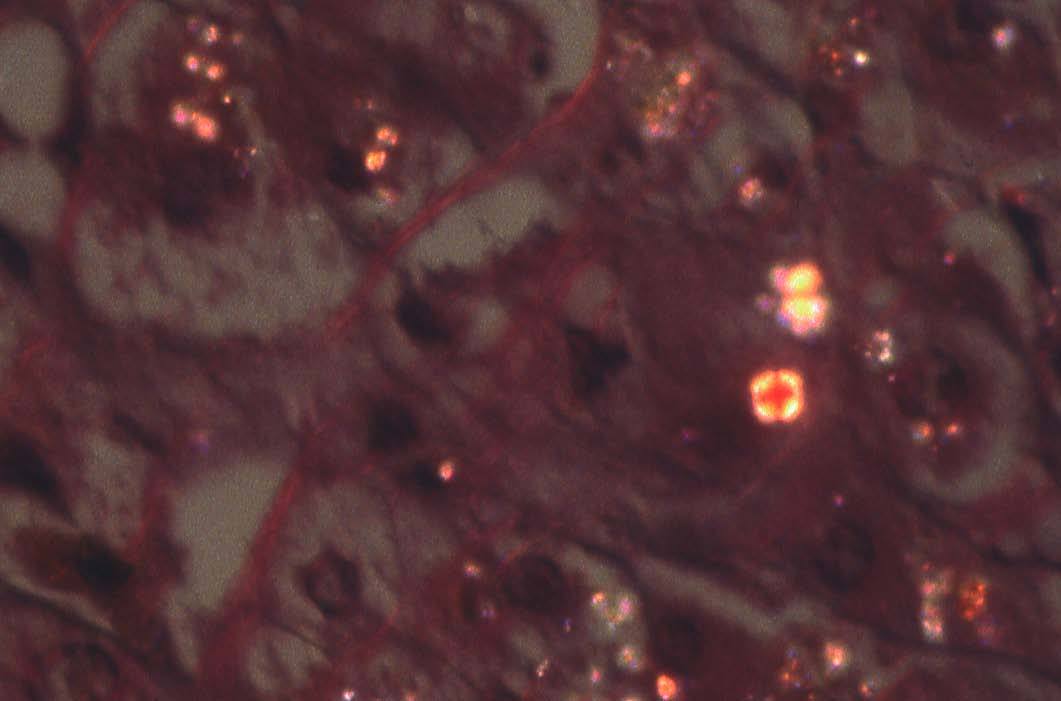
Fig. 2. Los depósitos de protoporfirina de mayor volumen adoptan el aspecto de cruz de Malta al exponerse a la luz polarizada; los de menor tamaño aparecen con un aspecto puntiforme. (Hematoxilina-eosina, *240.)
Con la biopsia hepática no es posible hacer una estimación pronóstica, ya que no permite determinar qué pacientes presentarán cirrosis hepática, ni cuántos con cirrosis desarrollarán un cuadro de insuficiencia hepática fulminante, aunque los casos con depósito intrahepático de PP sin cirrosis tienen mejor pronóstico que aquéllos con cirrosis. Los casos con cirrosis presentan, además de los depósitos de PP, cambios que apuntan a un patrón biliar, con proliferación colangiolar en los septos fibrosos.
TRATAMIENTO DE LA ENFERMEDAD HEPÁTICA
Para prevenir la aparición de complicaciones hepáticas graves, en los pacientes con PPE en quienes se detecta una afectación hepática leve se han ensayado varios tratamientos encaminados a reducir la carga de PP que recibe el hígado, aunque su utilidad no se ha demostrado mediante estudios controlados. Estos tratamientos consisten en: a) interrumpir la reabsorción ileal de PP; b) frenar la eritropoyesis y reducir de este modo la síntesis de PP, y c) facilitar la secreción biliar de PP.
La resincolestiramina es un quelante de las sales biliares que impide la reabsorción intestinal de la PP y promueve su eliminación fecal. Se ha utilizado en algunos casos a la dosis de 12-16 g/día por vía oral, con resultados excelentes16,17. También se ha empleado carbón activado (charcoal) para suprimir la reabsorción ileal de la PP que ha alcanzado el intestino. La dosis y frecuencia de administración del carbón activado (30 g/6 h) son empíricas y están condicionadas por la tolerancia del paciente18.
La supresión de la eritropoyesis se ha intentado mediante transfusiones repetidas19,20 y con la administración intravenosa de hematina21. La hematina (o arginato de hematina) es un producto purificado de hematíes humanos, comercializado en España con el nombre de Normosang® (laboratorios Orphan). Se administra por vía intravenosa a la dosis de 3-4 mg/día disuelta en seroalbúmina al 5% durante 4 días.
Pirlich et al22 consiguieron, con la administración de ácido ursodesoxicólico a la dosis de 18 mg/kg de peso/día a una paciente con PPE que presentaba ictericia y afectación importante de la función hepática, una mejoría bioquímica a las 2 semanas y normalización analítica a los 6 meses, que se atribuyó a la reducción de la citotoxicidad de una bilis rica en PP causada por el ácido ursodesoxicólico. Otros casos se han tratado con este fármaco23,24.
En los pacientes que han presentado ictericia y signos de insuficiencia hepática debe recurrirse al trasplante hepático, que, aunque no es curativo de la PPE, ya que los pacientes continúan presentando fotosensibilidad, permite salvar la vida de quienes han presentado esta complicación.
Mientras se está a la espera de proceder al trasplante conviene aplicar medidas terapéuticas, que en algunos pacientes han sido eficaces, como podría ser la exanguinotransfusión con hipertransfusión combinada con la administración de colestiramina. Gorchein y Foster25 administraron carbón activado a un paciente con insuficiencia hepatocelular y consiguieron una regresión notable de la enfermedad hepática y de la fotosensibilidad cutánea. Sin embargo, en otros casos, con este tratamiento sólo se logró una mejoría de la enfermedad cutánea, pero no de la hepática26,27.
Trasplante hepático
Hasta el momento se han efectuado unos 40 trasplantes hepáticos en pacientes con PPE12,28-43. La mayoría de pacientes presentaron una buena evolución después del trasplante. Se comprobó que la concentración de PP en los hematíes disminuía rápidamente y aumentaba la excreción fecal, y demostró la eficacia del aclaramiento hepático de PP por el injerto hepático44. Sin embargo, en los casos trasplantados persiste la fotosensibilidad y es posible la recurrencia del daño hepático en un plazo más o menos largo, por la continua llegada al hígado trasplantado de elevadas cantidades de PP45.
Se ha utilizado con éxito la administración de hematina intravenosa en un paciente con PPE que presentó lesión hepática grave a los 2 años de un trasplante35, así como las plasmaféresis repetidas asociadas a perfusión de albúmina en otro36.
Complicaciones del trasplante hepático
La cirugía del trasplante expone al paciente a riesgos derivados de la exposición durante la operación a las brillantes luces de las lámparas del quirófano, como son una neuropatía motora generalizada37,41, hemólisis intra y postoperatoria o quemaduras de la pared abdominal, que pueden causar la dehiscencia de la herida operatoria y perforaciones intestinales. Estas complicaciones se atribuyen a la activación de la PP del plasma, por lo que es esencial la aplicación de medidas para prevenir su aparición. La más importante es la utilización de filtros de acrilato amarillo en las lámparas a fin de reducir la exposición a la luz durante el acto quirúrgico11,20. La práctica de una exanguinotransfusión inmediatamente antes de la cirugía podría reducir de manera momentánea la fuente de PP y disminuir el riesgo de lesiones de fotosensibilidad. Esta medida podría sustituirse por plasmaféresis seguida de transfusiones sanguíneas.
El síndrome neurológico que se ha descrito en pacientes con PPE trasplantados, especialmente en quienes presentaban una colestasis más acentuada, cursa con dolor abdominal y en las extremidades muy intenso, resistente a veces a los opiáceos, una extrema debilidad muscular, incluso de los músculos intercostales, lo que puede provocar insuficiencia ventilatoria, y afectación de los pares craneales. El electromiograma muestra una degeneración axonal de las fibras motoras distales. Suele manifestarse inmediatamente después del trasplante y se atribuye a un efecto neurotóxico de la PP plasmática11,12. Algunos pacientes experimentan una mejoría, pero otros no.
Tratamiento de la litiasis
La litiasis vesicular sólo tiene indicación quirúrgica si ocasiona alguna de sus complicaciones, cólicos hepáticos, obstrucción biliar o colecistitis. La cirugía debe efectuarse en quirófanos con protección frente a la luz quirúrgica atenuada con filtros de acrilato amarillo.
MONITORIZACIÓN DEL DAÑO HEPÁTICO EN LOS PACIENTES CON PROTOPORFIRIA ERITROPOYÉTICA
Por desgracia no disponemos de criterios para detectar precozmente los casos que pueden desarrollar un cuadro de insuficiencia hepática. Sin embargo, el hecho de que todos los pacientes que presentan insuficiencia hepática hayan sido diagnosticados de enfermedad hepática con anterioridad justifica que se siga con atención a los pacientes con signos de alteración hepática, aunque debe tenerse en cuenta que el proceso tiene un curso insidioso y, cuando se constata un cuadro clínico de lesión hepática importante, se ha llegado al final del proceso.
Se ha señalado que una concentración de PP en eritrocitos superior a 1.500 µg/dl y una excreción fecal inferior a 50 µg/dl representan un riesgo de enfermedad hepática grave. La ratio entre la concentración de PP fecal y eritrocitaria, que oscila entre 0,1 y 1,1, es inferior a 0,05 en los pacientes con insuficiencia hepática. Desgraciadamente no se dispone de estudios longitudinales para conocer qué valores permiten establecer un diagnóstico precoz. Se ha indicado que el aumento de la excreción urinaria de coproporfirinas, con un cambio en la proporción de los isómeros I y III, es un indicador preciso y temprano de la afectación hepática de la PPE, de modo que el daño hepático se asociaría a un predominio del isómero I respecto al III7.
Los pacientes con PPE deben evitar el consumo excesivo de alcohol, la toma de fármacos que pueden inducir colestasis, como los estrógenos, y la restricción calórica, ya que inhibe la acción de las enzimas y, por tanto, puede exacerbar la PPE. En caso de episodio de litiasis coledocal, debe procederse a la liberación del obstáculo lo más rápidamente posible, a poder ser por vía endoscópica para evitar la exposición lumínica del quirófano.
Conviene que periódicamente (p. ej., una vez al año o cada 6 meses) se efectúen pruebas hepáticas y una ecografía abdominal a los pacientes con PPE para apreciar si hay o no colelitiasis. Si se comprueba una elevación enzimática, sería conveniente proceder a una punción-biopsia hepática y, si se demuestra la presencia de depósitos de PP, iniciar tratamiento con resincolestiramina o con ácido ursodesoxicólico.
El examen periódico de las concentraciones de PP en la sangre y en las heces puede facilitar el reconocimiento de un empeoramiento del estado del hígado, ya que en estos casos disminuye la excreción fecal de PP, mientras aumenta la concentración plasmática. Se debe informar a los pacientes de que la constatación de alguno de estos síntomas, como dolor abdominal, cansancio desproporcionado a la actividad física realizada, ictericia o mayor sensibilidad a la luz de la que tenían antes, exige una consulta médica. En caso de constatación de insuficiencia hepatocelular, se planteará la indicación de un trasplante hepático.

