







INTRODUCCIÓN
La enfermedad de Caroli (EC), o ectasia comunicante de las vías biliares intrahepáticas, es una enfermedad congénita caracterizada por múltiples dilataciones saculares o quísticas de las vías biliares intrahepáticas. Fue descrita por primera vez por Caroli et al1, en 1958, como una entidad clínica consistente en colangitis, litiasis intrahepática y abscesos hepáticos. En 1964, el propio Caroli, junto con Corcos, extendió sus observaciones acerca de esta enfermedad indicando la existencia de 2 tipos distintos: uno en el que las dilataciones saculares de las vías biliares intrahepáticas se asocian a fibrosis hepática congénita, que cursa clínicamente con hipertensión portal, y otro sin fibrosis2. Algunos autores utilizan la denominación enfermedad de Caroli cuando las dilataciones biliares son la única anomalía detectable y síndrome de Caroli cuando se asocia fibrosis hepática congénita.
ETIOPATOGENIA
La EC es el resultado de un defecto en la remodelación de la placa ductal en la fase de la embriogénesis en que se desarrollan los conductos biliares intrahepáticos de mayor calibre. Esto se asocia, en grado variable, a persistencia de las estructuras biliares embrionarias, fibrosis y dilatación ductal. El remodelado de la placa ductal empieza en el hilio y se extiende a la periferia, por lo que hay un espectro de alteraciones morfológicas que depende del nivel en el que se altera el desarrollo embrionario3. La EC es el resultado de la afección de los conductos intrahepáticos mayores, y el síndrome de Caroli aparece cuando, además, están involucrados los pequeños conductos interlobulillares, lo que causa una fibrosis hepática que coexiste con la dilatación quística de las vías biliares intrahepáticas característica de la EC.
Aunque se cree que el quiste de colédoco se origina por una anomalía de la unión biliopancreática y no de la placa ductal, esta malformación puede presentarse en asociación con la EC4. La placa ductal y la diferenciación tubular renal comparten determinantes genéticos, por lo que no es infrecuente la asociación de EC con riñón poliquístico o ectasia tubular renal5. Como la forma habitual de poliquistosis renal, la EC se transmite con una herencia autosómica recesiva. Sin embargo, del mismo modo que existe una forma de poliquistosis renal de transmisión autosómica dominante, en algunos casos de EC también hay una penetrancia familiar que sugiere una herencia dominante.
A pesar de las claras evidencias de que la EC tiene una base genética, no se han llevado a cabo estudios específicos de detección de los defectos moleculares que pudieran explicarla. En los casos asociados con poliquistosis renal autosómica recesiva, puede estar involucrado el defecto molecular de la enfermedad renal, causada por mutaciones del gen PKHD1, que codifica para la fibrocistina, una proteína estructural de los cilios. Las alteraciones de la fibrocistina causan disfunción ciliar, que se considera el principal mecanismo patogénico de la cistogénesis. La proliferación celular excesiva que caracteriza el epitelio biliar quístico se asocia a una disregulación del factor de crecimiento epidérmico (EGF) y probablemente también a la de los receptores estrogénicos6. Se desconoce si esta última anomalía puede estar involucrada en la asociación de EC con colestasis del embarazo, más frecuente de lo que podría esperarse, por la escasa frecuencia de ambas enfermedades en la población.
La EC podría estar relacionada con una mutación del gen ABC B4 (ATP-binding casette), denominado también MDR3 (multidrug-resistance-3), que codifica para una proteína cuya función es el transporte activo de fosfatidilcolina (lecitina) a través de la membrana canalicular de los hepatocitos7. Su déficit determina la ausencia de fosfolípidos en la bilis, lo cual acelera la precipitación del colesterol biliar, y la formación de barro biliar y cálculos tanto en la vesícula como en los conductos biliares8. Se han encontrado defectos del gen MDR3 en un subtipo de colestasis intrahepática familiar, en algunos casos de colestasis del embarazo, y en algunos pacientes con litiasis biliar de colesterol9.
Con respecto a las mutaciones del MDR3 y la litiasis biliar, de modo notable Rosmorduc et al10 describieron a 6 pacientes con mutaciones del gen MDR3 en heterocigosis que cursaban con colestasis y barro o microcálculos en las vías biliares intrahepáticas, cuya bilis estaba sobresaturada con colesterol, en parte por una profunda deficiencia de fosfolípidos biliares8. Lucena et al11 describieron a una mujer con una mutación MDR3 y cirrosis biliar con antecedentes de colelitiasis en la juventud y colestasis del embarazo. Aunque en estos casos no se mencionan defectos de las vías biliares intrahepáticas, la presentación clínica con litiasis y barro intrahepático es característica de pacientes en fases tempranas de la EC12, por lo que es plausible que la deficiencia de MDR3 esté involucrada en la hepatolitiasis típica de esta enfermedad. Otra evidencia a favor de esta etiología es la observación de deficiencia de fosfolípidos en la bilis hepática y reducción del ARNm de MDR3 en el tejido hepático en una serie japonesa de pacientes sometidos a resección parcial hepática por hepatolitiasis13. Los estudios genómicos, aún pendientes, probablemente contribuirán a un mejor conocimiento de la etiopatogenia de la EC.
MANIFESTACIONES CLÍNICAS
En la EC, las dilataciones quísticas del árbol biliar dificultan el flujo normal de la bilis, cuya estasis facilita la precipitación de sólidos. Si la estasis biliar coexiste con una bilis sobresaturada con colesterol, precipitan cristales de colesterol, mientras que en presencia de infección bacteriana se forman precipitados amorfos de bilirrubinato cálcico14. En fases tempranas de la enfermedad, los cálculos presentes en las vías biliares intrahepáticas suelen ser de colesterol, como se demuestra indirectamente por el frecuente hallazgo de numerosos cristales de colesterol en el examen microscópico del sedimento de la bilis obtenida por intubación duodenal12. En cualquier caso, es muy frecuente la formación de barro biliar y cálculos intrahepáticos, de colesterol, pigmentarios o mixtos, que son, en gran parte, los causantes de la semiología de obstrucción biliar.
La manifestación habitual de la EC es el dolor abdominal en forma de crisis de presentación recurrente, usualmente debido a la litiasis intrahepática, a veces a litiasis vesicular asociada, y otras a episodios de pancreatitis aguda, por paso de cálculos intrahepáticos a la vía biliar principal y enclavamiento en la unión coledocopancreática12. Obviamente, en estos casos puede haber ictericia de intensidad variable.
Otra manifestación frecuente de la EC son las crisis de colangitis, que pueden conducir a la formación de abscesos intrahepáticos. La colangitis puede ser de aparición espontánea, pero con más frecuencia aparece después de exámenes cruentos del árbol biliar, como la colangiografía retrógrada endoscópica, o intervenciones quirúrgicas, como hepatoyeyunostomía o coledocoyeyunostomía, que han permitido la entrada de gérmenes intestinales en el interior del árbol biliar. Usualmente, la colangitis está causada por gérmenes gramnegativos15.
Los pacientes con EC asociada a fibrosis hepática congénita pueden presentar algunas de las consecuencias de la hipertensión portal, como una hemorragia digestiva por rotura de varices esofágicas.
Los síntomas de la enfermedad pueden aparecer durante la infancia o en la edad adulta; incluso se ha descrito a 2 pacientes que se diagnosticaron en la séptima y la octava décadas de la vida, respectivamente16,17. Generalmente, el diagnóstico de la causa de estos síntomas suele hacerse años después del inicio del cuadro clínico. El examen físico suele mostrar hepatomegalia. Si existe fibrosis hepática congénita, es frecuente detectar una esplenomegalia como consecuencia de la hipertensión portal. En las pruebas hepáticas, la única anomalía suele ser la elevación de las enzimas de colestasis (gammaglutamil transpeptidasa [GGT] y fosfatasa alcalina [FA]), con aumentos, en general, poco marcados de la aspartato aminotransferasa (AST) y la alanina aminotransferasa (ALT).
DIAGNÓSTICO
El diagnóstico de la EC es esencialmente radiológico (revisado en Levy y Bohrmann18). La observación, en una ecografía abdominal o en una tomografía computarizada, de imágenes de áreas quísticas dentro del hígado, especialmente si contienen imágenes de litiasis en su interior, es altamente sospechosa19,20 (fig. 1). Para demostrar que las cavidades quísticas están comunicadas con el árbol biliar puede efectuarse una colangiografía por resonancia magnética (fig. 2) o una gammagrafia hepática con derivados del ácido iminodiacético21. Con fines exclusivamente diagnósticos, no debe efectuarse una colangiografía retrógrada endoscópica, que sería la exploración más directa, para evitar la contaminación bacteriana de las vías biliares, difícil de erradicar en presencia de zonas con estasis biliar.
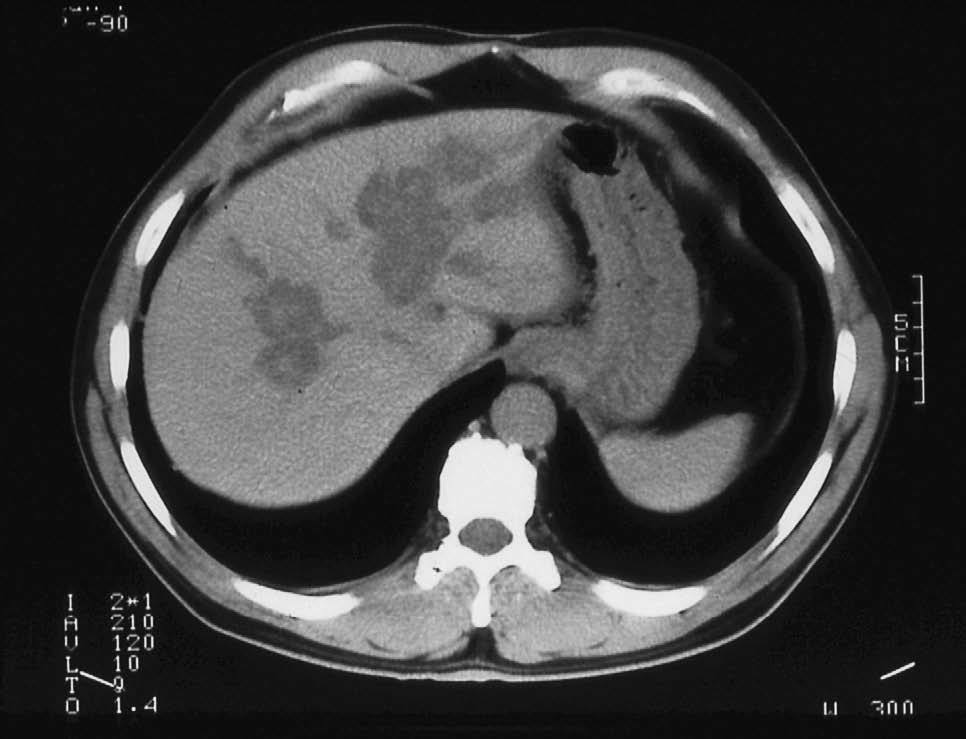
Fig. 1. Tomografía computarizada. Imágenes quísticas intrahepáticas con litiasis en el interior de las cavidades quísticas.
De acuerdo con la topografía de las dilataciones de las vías biliares, la EC puede clasificarse en difusa, cuando afecta los conductos biliares de ambos lóbulos hepáticos, o monolobular, si se localizan en un solo lóbulo hepático, generalmente el izquierdo, lo que ocurre en, al menos, una cuarta parte de los casos16. Las vías biliares que se afectan preferentemente son los conductos segmentarios, por lo que no es previsible obtener una representación de éstos si efectúa una punción-biopsia hepática, que sólo será informativa en caso de EC asociada a fibrosis hepática congénita.
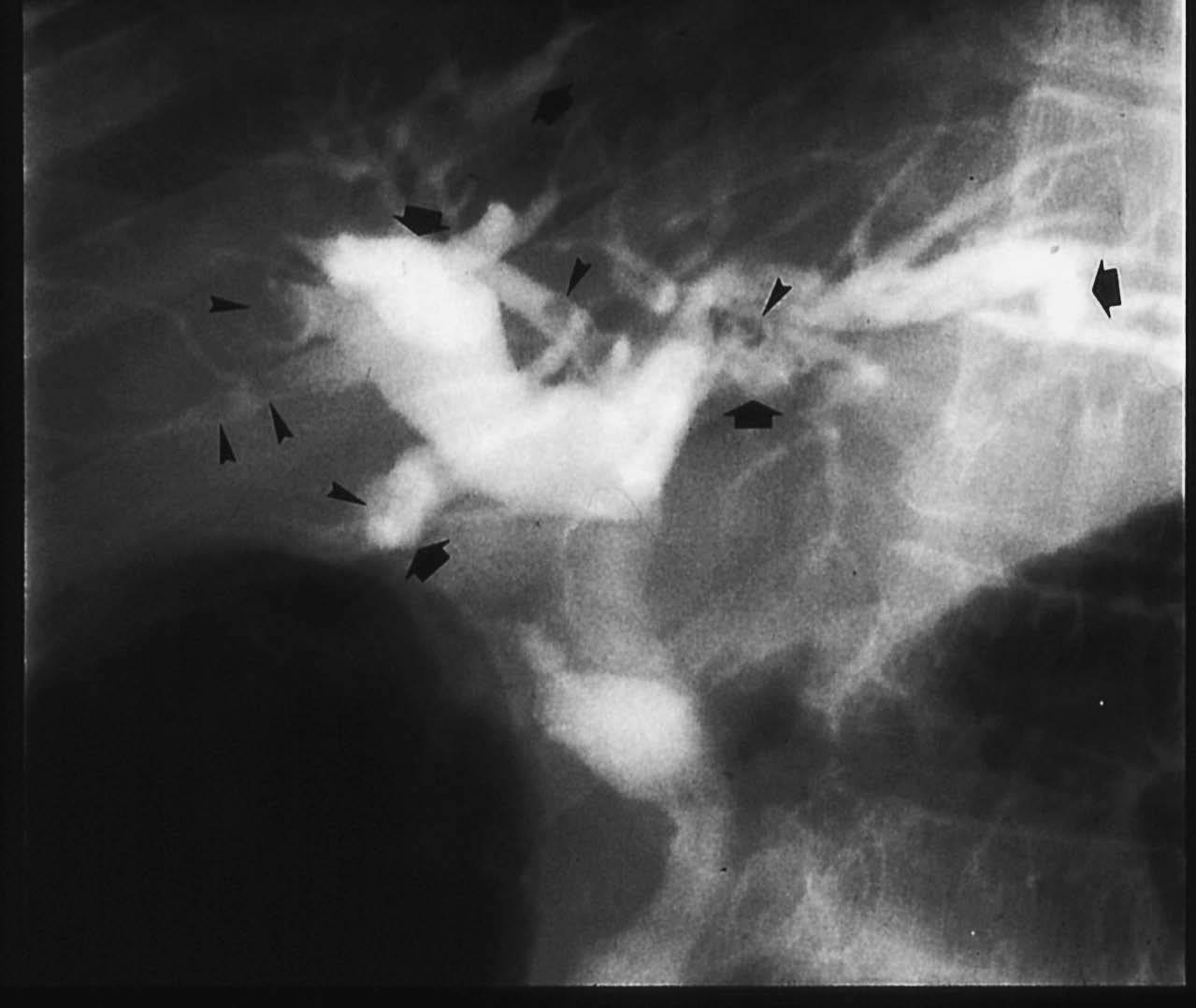
Fig. 2. Colangiografía retrógrada. Litiasis intrahepáticas (cabezas de flecha) en un árbol biliar con dilataciones saculares.
Para sospechar el diagnóstico de EC es muy importante valorar la existencia de imágenes de litiasis intrahepáti ca en la ecografía abdominal, dispuestas de forma lineal (fig. 3), lo que sugiere que están situadas en el interior de la vía biliar. Estas imágenes se observan en pacientes en los que no se distinguen imágenes quísticas, porque las dilataciones de las vías biliares son de pequeño tamaño. No pocas veces estas imágenes ecográficas son mal interpretadas como granulomas calcificados; sin embargo, la presencia de dolor biliar y/o colestasis anictérica es altamente sospechosa de que existan cálculos intrahepáticos.
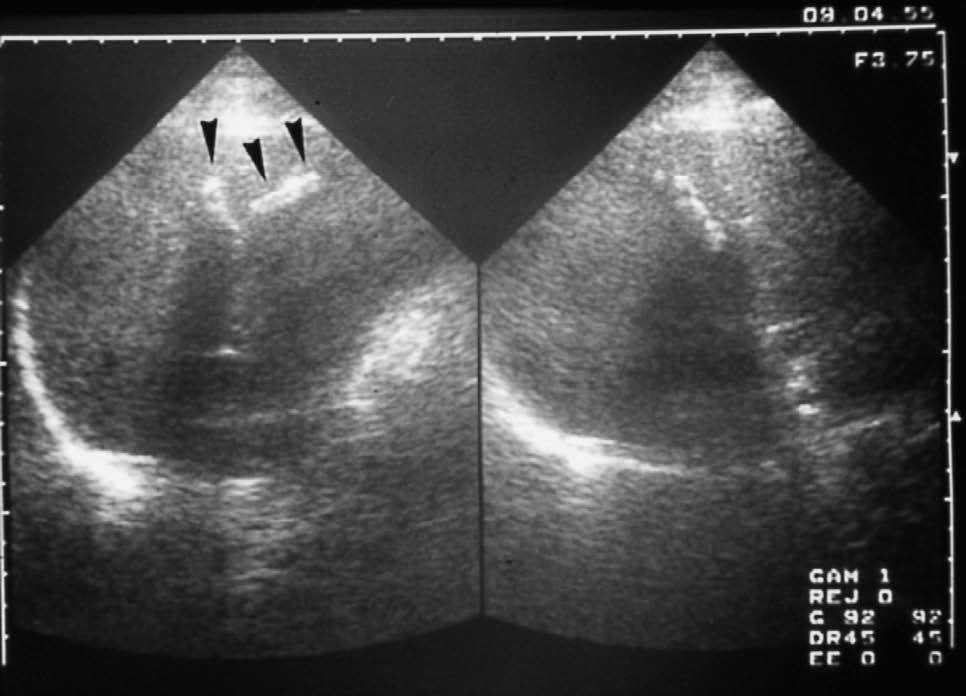
Fig. 3. Ecografía abdominal. Litiasis intrahepáticas con disposición lineal (flechas).
HISTORIA NATURAL
Los pacientes con colangitis recurrente rebelde al tratamiento antibiótico pueden fallecer por una sepsis o por complicaciones de una amiloidosis secundaria22. Los pacientes con EC pueden desarrollar un colangiocarcinoma, complicación temible que afecta a entre el 7 y el 14% de los casos23,24 y se atribuye al efecto carcinogénico de la exposición repetida del epitelio ductal a concentraciones elevadas de ácidos biliares secundarios y desconjugados por la acción bacteriana25. Por desgracia, no disponemos de ningún método para detectar el desarrollo de un colangiocarcinoma en una etapa inicial que pudiera permitir la indicación de un trasplante hepático. En nuestra experiencia, el tratamiento ininterrumpido con ácido ursodesoxicólico (AUDC), en casos tempranos12, o el trasplante ortotópico de hígado, en casos avanzados26-28, cambian favorablemente la historia natural de la enfermedad.
TRATAMIENTO
El objetivo fundamental del tratamiento consiste en prevenir la aparición de colangitis o tratar eficazmente los casos que ya hayan presentado esta complicación. Otros objetivos importantes son evitar los ataques de dolor biliar y/o pancreatitis derivados del paso de cálculos al colédoco y la subsiguiente obstrucción de la vía biliar principal o pancreática.
Tratamiento de la colangitis
En los pacientes con EC monolobular, la mejor decisión es la hepatectomía parcial, con la consiguiente eliminación de los segmentos patológicos del árbol biliar y la resolución definitiva del problema, tanto la infección como el riesgo de desarrollar un colangiocarcinoma16,28. En los casos con afección diseminada deben utilizarse antibióticos, betalactámicos, aminoglucósidos y quinolonas. Si la fiebre reaparece después de abandonar el tratamiento antibiótico, hecho nada infrecuente, debe procederse a un tratamiento continuado y pensar en alguna actuación más radical.
Las medidas para conseguir un drenaje de la bilis retenida e infectada han sido eficaces en pocos casos. Las intervenciones quirúrgicas habitualmente no consiguen un drenaje eficaz y tienen una alta mortalidad. En algún paciente con afección bilobular se ha efectuado, con éxito, una resección parcial de las áreas más afectadas29. El trasplante hepático se ha sido utilizado con éxito en pacientes con colangitis recurrente y, probablemente, es el tratamiento de elección en esta situación26-28.
Tratamiento del dolor abdominal
Si los episodios dolorosos remedan los cólicos biliares típicos y existe litiasis vesicular, debe considerarse la conveniencia de efectuar una colecistectomía laparoscópica. Cuando la vesícula no está ocupada y existe hepatolitiasis, es preferible ensayar la administración de AUDC en dosis de 15-20 mg/kg/día. Ros et al12 demostraron que la bilis de una elevada proporción de pacientes con EC presenta una sobresaturación de colesterol, lo que justifica este tratamiento, que ejerce, además de un efecto disolutivo de la litiasis de colesterol, un aumento del flujo biliar que facilita la movilización de los microcristales y del barro biliar presente en las áreas dilatadas. En pacientes con EC poco avanzada pero con litiasis intrahepática sintomática, el tratamiento prolongado con AUDC puede detener el crecimiento de los cálculos o disolverlos, reducir la frecuencia de los cólicos biliares y normalizar las alteraciones bioquímicas hepáticas. Nuestra experiencia con 20 pacientes portadores de una EC poco avanzada, que comenzaron con un dolor abdominal o pancreatitis por movilización de cálculos intrahepáticos de pequeño tamaño y con un seguimiento de 6 a 20 años, demuestra que el tratamiento ininterrumpido con AUDC a dosis iniciales de 20 mg/kg y de mantenimiento de 10 mg/kg tiene una notable eficacia para erradicar los síntomas de dolor biliar o los ataques de pancreatitis, prevenir episodios de colangitis, mejorar o normalizar las pruebas de función hepática y, en definitiva, detener la evolución de la enfermedad y evitar, quizá, el desarrollo de un colangiocarcinoma. La litiasis intrahepática también puede tratarse mediante litotricia colangioscópica percutánea transhepática, si se dispone de esta técnica superespecializada.
Tratamiento de la obstrucción biliar
Si ha ocurrido una obstrucción del colédoco por litiasis debe intentarse una esfinterotomía endoscópica con extracción de los cálculos30. En caso de que el paciente no hubiera presentado con anterioridad crisis de colangitis, la técnica debe efectuarse con cobertura antibiótica para minimizar la introducción de una infección en la vía biliar31. La recidiva de coledocolitiasis obstructiva en pacientes con esfinterotomía previa puede tratarse, con buenas posibilidades de éxito, mediante litotricia con ondas de choque, evitando así la manipulación de la vía biliar y el subsiguiente riesgo de infección.
Tratamiento de la hipertensión portal
Los pacientes con EC y fibrosis hepática congénita están expuestos a sangrar por una rotura de las varices esofágicas. Si esto ocurre, deben aplicarse las medidas habituales de hemostasia primaria, con reposición de la volemia y administración de somatostatina. En caso de hemorragia recidivante, hay que plantearse la opción de colocar un cortocircuito portosistémico intrahepático transyugular (TIPS) o efectuar una derivación portosistémica quirúrgica, que no comporta el riesgo de inducir una encefalopatía portocava, como ocurre en los pacientes cirróticos ya, que, en estos pacientes, la función hepática suele estar conservada. En los pacientes con antecedente de hemorragia por varices, debe proponerse la prevención secundaria del sangrado mediante bloqueadores beta y mononitrato de isosorbida.
Tratamiento de los casos asintomáticos
En los individuos en quienes se ha efectuado el diagnóstico de EC en el curso de la exploración de una colestasis asintomática, puede ensayarse la administración profiláctica de AUDC, que suele mejorar e incluso normalizar las pruebas hepáticas. En caso contrario, puede asumirse que el tratamiento es ineficaz y abandonarlo, para seguir con controles clínicos y ecográficos periódicos.
CONCLUSIÓN
La EC es una enfermedad biliar de probable transmisión autosómica recesiva, de presentación infrecuente pero no excepcional. Su origen se encuentra en defectos embrionarios en la placa ductal, que conducen a dilataciones saculares de las vías biliares intrahepáticas, asociadas o no a fibrosis hepática congénita, y coexisten con frecuencia con otras anomalías genéticas biliares o renales. Se desconocen sus bases moleculares. Cursa con colestasis biológica asintomática o con dolor abdominal, colangitis y obstrucción biliar, debida a hepatolitiasis. El diagnóstico es radiológico: puede sospecharse por los hallazgos de la ecografía o tomografía computarizada y se confirma mediante colan giografía por resonancia magnética. La enfermedad de larga evolución puede complicarse con el desarrollo de un colangiocarcinoma, de pronóstico infausto. Una vez establecida la colangitis, es muy difícil erradicarla con tratamiento antibiótico que, a lo sumo, controlará temporalmente la sepsis biliar. El drenaje quirúrgico de los segmentos biliares afectados puede ser útil en casos de colangitis intratable, pero la curación definitiva sólo es posible mediante lobectomía, en pacientes con enfermedad limitada a un lóbulo hepático, o con trasplante hepático, en los casos de enfermedad difusa. El tratamiento con dosis altas de AUDC en pacientes con enfermedad poco avanzada y hepatolitiasis de pequeño tamaño es altamente beneficioso, pues detiene la evolución de la enfermedad y evita sus complicaciones.

