









El sarcoma histiocítico (SH) es una neoplasia maligna rara, que se origina de células tallo mieloides de médula ósea, con características morfológicas e inmohistoquímicas de histiocitos (macrófagos) maduros. El SH se puede presentar en ganglios linfáticos y en sitios extraganglionares como la piel, tejidos blandos y particularmente el aparato gastrointestinal, por lo general asociado con enfermedad avanzada y curso clínico agresivo. El diagnóstico de SH recae en la presencia de las características morfológicas e inmunohistoquímicas de estirpe histiocítica, y la exclusión de linfoma de células grandes y otras neoplasias poco diferenciadas como carcinomas y melanomas
Histiocytic sarcoma (HS) is a rare malignant tumor, originating from bone marrow-derived myeloid stem cells, showing morphologic and immunophenotipic evidence of mature tissue histiocytes (macrophages). HS arises in lymph nodes and extranodal sites such as skin, soft tissues and particularly the gastrointestinal tract, often presenting with clinically advanced disease and pursuing and aggressive clinical course. The diagnosis of HS relies predominantly on the immunohistochemical features of the histiocytic lineage, and the exclusion of large cell lymphoma and other poorly differentiated large cell malignancies such as, carcinomas and melanomas.
¿ INTRODUCCIÓN
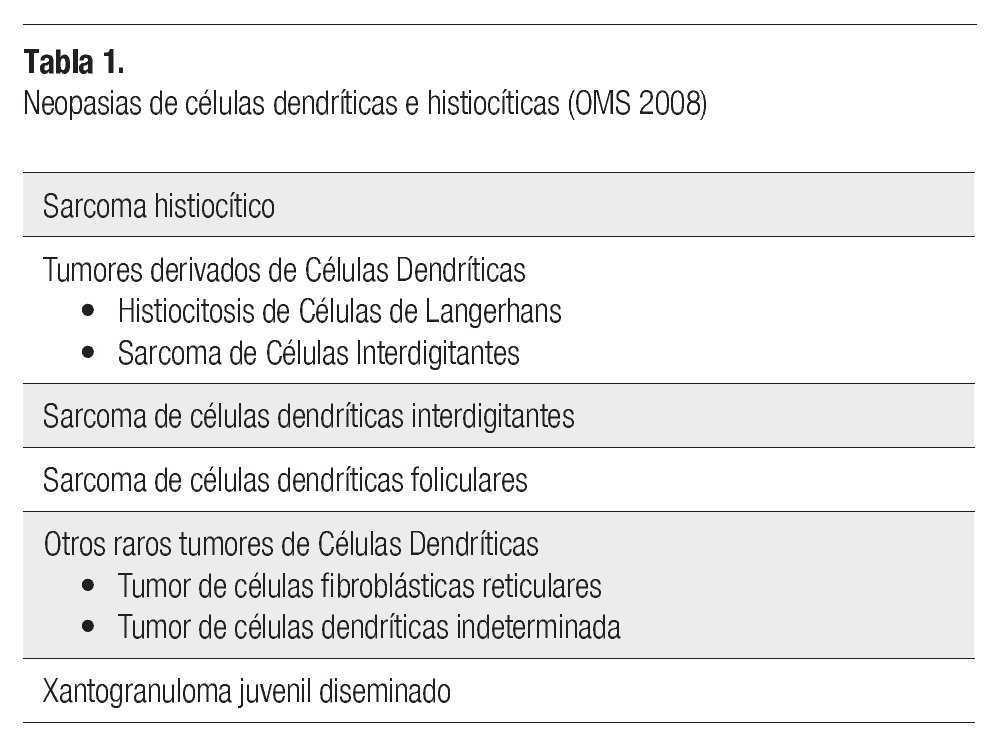
Los macrófagos (también llamados histiocitos), son células del sistema fagocítico mononuclear, cuyo citoplasma contiene numerosos lisosomas y su función es la fagocitosis.1 De la misma familia, derivadas de una célula madre mieloide, están las células dendríticas, cuya función es la presentación de antígenos y en este grupo se incluyen las células de Langerhans, las células dendríticas interdigitantes, las células dendríticas reticulares y las células dendríticas plasmocitoides.2 Las neoplasias de células dendríticas/histiocíticas se agrupan según la Organización Mundial de la salud en: sarcoma histiocítico (SH), tumores derivados de células de Langerhans, sarcoma de células dendríticas interdigitantes, sarcoma de células dendríticas foliculares, xantogranuloma juvenil diseminado y otras neoplasias de células dendríticas raros (si otra especificación) (Tabla 1).
El SH (previamente designado linfoma histiocítico verdadero) es una neoplasia maligna, de etiología desconocida, cuyas células presentan características morfológicas e inmunofenotípicas de macrófagos maduros (histiocitos) y representa menos de 1% de todas las neoplasias hematológicas.3-7 Históricamente, estos tumores se han incluidos bajo diferentes términos como el de reticulosarcoma (retothelsarkom) de Roulet y Oberling,8 reticulosis medular histiocitaria/histiocitosis maligna de Scott y Robb Smith, o "linfomas histiocíticos verdaderos", como fueron designados por Henry Rappaport en 1966.9-14 (Figura 1). Es muy probable que esta variedad de términos aplicados a una sola entidad, se base en el hecho de que los autores mencionados, establecieron sus diagnósticos únicamente en cortes teñidos con hematoxilina y eosina. Muchos de los tumores que originalmente se diagnosticaron como SH con base en esta técnica, fueron reclasificados como linfomas T, linfomas anaplásicos de células grandes, linfomas NK/T, linfomas inmunoblásticos, linfoma de Hodgkin clásico, carcinomas, melanomas o diversos sarcomas, cuando estuvieron disponibles nuevos métodos como la inmunohistoquímica y técnicas de biología molecular.4,15-16

Figura 1. Henry Rapapport A), Charles Oberling B) y Alastair Robb-Smith C).
El término "sarcoma histiocítico" fue propuesto originalmente por Georges Mathé y colaboradores del Instituto Gustave-Roussy, en París en el año de 1970,17 (Figura 2). La Organización Mundial de la Salud en su clasificación del 2008, define al SH como una neoplasia maligna con características morfológicas e inmunofenotípicas, semejantes a los histiocitos maduros.4 Aproximadamente 50% de los casos se presentan en ganglios linfáticos, el resto en sitios extraganglionares, afectando principalmente el aparato digestivo, la piel y el sistema nervioso central.6,18,19 El diagnóstico de SH se basa en la verificación de linaje histiocítico, por medio de inmunohistoquímica y la exclusión de otras neoplasias malignas de células grandes poco diferenciadas.3

Figura 2. Georges Mathé y la portada del artículo donde se da el nombre de Sarcomas Histiocítico. British Journal of Cancer 1970;24:687-695.
¿ MONOPOYESIS Y DIFERENCIACIÓN HACIA MOCRÓFAGOS
Fue el histólogo francés Louis-Antoine Ranvier quien denominó como "clasmocitos" (clasmocitos de Ranvier) (que significa células que se fragmentan, porque supuso que sus cortas prolongaciones podrían separarse del cuerpo celular), a las células con capacidades fagociticas. Reconoció que estos "clasmocitos" se transformaban en elementos con capacidad fagocítica, cuando algunas partículas extrañas entraban al organismo. Posteriormente, Joseph Louis Renaut observó que algunos clasmocitos contenían granulaciones intracitoplásmicas, como ya lo había indicado Ranvier, y pensó que tales elementos tenían la capacidad de elaborar productos de secreción y las llamó "células ragiocrinas" (que producen elementos celulares migrantes). La fagocitosis fue reconocida en 1880 por Elie Metchnikoff, por lo que fue galardonado con el premio Nobel en medicina en 1908 (compartido con Paul Ehrlich).20 Aunque William Osler informó cinco años antes (en 1875), en un análisis histológico de pulmones de mineros, la fagocitosis de partículas de carbón e inyectó tinta china en las axilas y pulmones de gatos, donde histológicamente encontró células cargadas con gránulos oscuros.21 El término de fagocitosis fue utilizado por Metchikoff hasta 1884,21 Alexander A. Maximow determinó la morfología y el papel funcional de los "clasmocitos de Ranvier" y los llamó poliblastos (Poliblastos de Maximow). Él observó que estas células contenían numerosos granulaciones intracitoplásmicas, y supuso que eran productos de secreción. Metchnikoff les llamó macrófagos para diferenciarlos de los micrófagos (como fueron conocidos los polimorfonucleares neutrófilos originalmente), y esta capacidad fagocítica, fue lo que dio idea a Ludwig Aschoff para proponer el concepto de "sistema retículo endotelial", que posteriormente fue llamado sistema fagocítico mononuclear por Ralph van Furth y colaboradores.22,23
Los macrófagos (histiocitos) se originan de monocitos procedentes de la médula ósea. A partir de una célula madre pluripotencial se originan dos línea celulares, la célula madre linfoide y célula madre mieloide (CMM). La CMM da lugar a la unidad formadora de colonias (UFC) de la serie basófila, eosinófila, eritrocítica, megacariocítica y una célula madre bipotencial para la serie granulocítica-monocítica, de esta última deriva la UFC para la serie neutrofila y monocítica, de la que proviene el monoblasto. El monoblasto es una células grande (15-25μ) que se caracteriza por tener citoplasma abundante basófilo, núcleo redondo y numerosos nucléolos, que posteriormente se diferencia hacia promonocito (15-20μ), con citoplasma intensamente basófilo (por su gran cantidad de polirribosomas) y con núcleo irregular formado por pliegues o surcos. Los promonocitos pueden proliferar rápidamente formando monocitos, que salen de la médula ósea hacia la sangre.1,24 Los monocitos cumplen su función en los diferentes tejidos donde se diferencian hacia macrófagos tisulares (histiocitos). La monopoyesis es estimulada por diversos factores de crecimiento que incluyen: GM-CSF (factor estimulante de colonias granulocito-monocito), M-CSF (factor estimulante de colonias de macrófagos), monocitopoyetina y la IL-3 (Interleucina 3).25 Se ha propuesto, que debido a que las los SH expresan características morfológicas e inmunofenotípicas de macrófagos, pudieran ser estas células el origen de estas neoplasias (Figura 3).
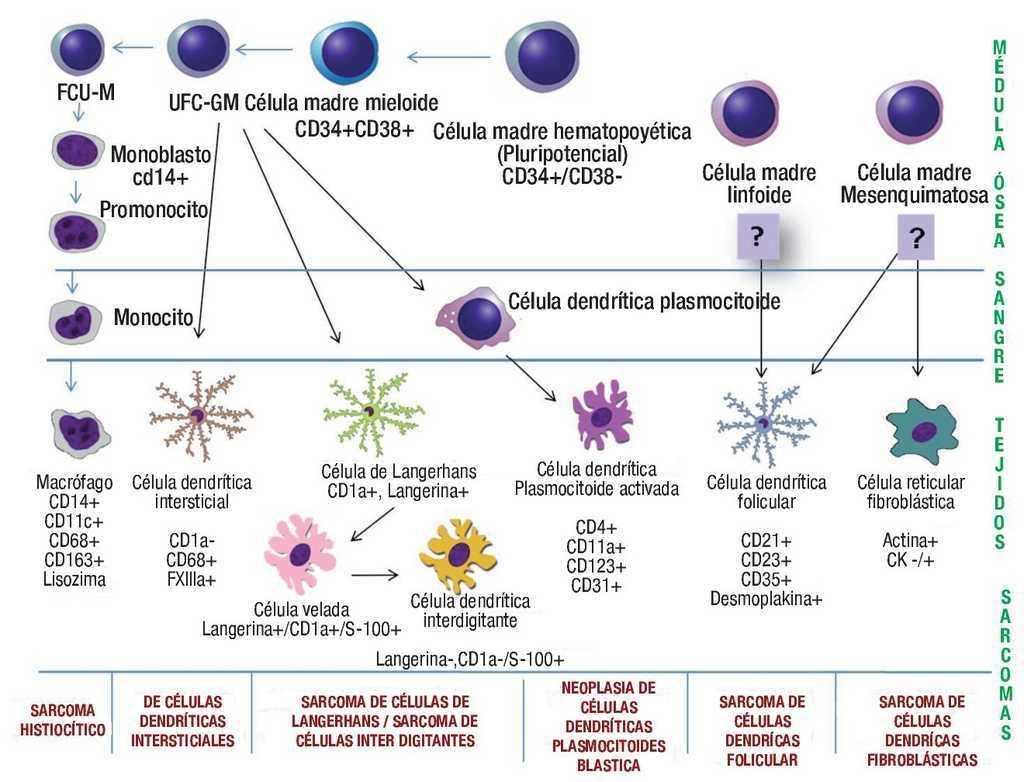
Figura 3. Esquema donde se representa la diferenciación de las células dendríticas/ histiocíticas, haciendo alusión a sus respectivas neoplasias.
Por otro lado, la maduración de células dendríticas es a partir de UFC granulocítica-monocítica que está regulada por la IL-4, el factor estimulante de colonias macrófago/granulocito y el factor de necrosis tumoral a. Existe una gran flexibilidad en este sistema, tanto macrófagos como células dendríticas pueden derivar en la dirección una de la otra, es decir, las células con fenotipo dendrítico pueden diferenciarse hacia un fenotipo de macrófagos por acción de IL-10 o M-CSF. Por lo anterior, no es de sorprender que exista una "zona gris", en la cual la distinción de macrófagos y de células dendríticas no sea absoluto, y ésta es la explicación del por qué algunas neoplasias histiocíticas/dendríticas pueden compartir inmunofenotipo.26
El origen del precursor de las células dendríticas foliculares aún no está claro. Hay autores que han sugerido un origen hematopoyético por la expresión de algunos antígenos asociados a células hematopoyéticas.27 No obstante, hay evidencias recientes de que derivan de células mesenquimatosas, que se ha hecho evidente tanto por la morfología fibroblásticas que presentan in-vitro, como por la expresión de antígenos asociados a los fibroblastos y por la falta de expresión de CD45 (antígenos lecocitario común).28
¿ PRESENTACIÓN CLÍNICA
Los SH se presentan desde la infancia hasta la vejez, sin embrago la mayoría de los casos ocurren en adultos, con una edad media de 52 años de edad.29 No hay predilección por el género, ya que varía según las series informadas.3,7,18 Las manifestaciones dependerán del sitio de afección (Tabla 1). Los pacientes pueden presentar síntomas sistémicos inespecíficos como fiebre, mal estado general, anorexia, astenia, pérdida de peso, pancitopenia y hepatoesplenomegalia. Cuando el SH afecta la piel, las manifestaciones pueden ser diversas e ir desde una erupción cutánea (rash), de apariencia benigna hasta lesiones solitarias nodulares o numerosos tumores, afectando tronco y extremidades. Los pacientes con lesiones intestinales a menudo manifiestan dolor, pesantez abdominal y obstrucción intestinal. Cuando hay afección ósea, suele haber dolor y en ocasiones pueden identificarse lesiones líticas por estudios de imagenología.30
Algunos casos se han presentado en pacientes con tumor de células germinales mediastinales (la mayoría teratoma maligno con o sin componente de tumor del saco vitelino). Se ha sugerido que a partir del teratocarcinoma, las células malignas pueden diferenciarse hacia células hematopoyéticas.31 Es de interés que los pacientes con tumores germinales no seminomatosos, tienen un riesgo mayor que el de la población general de desarrollar alteraciones hematológicas.32
El SH puede asociarse a linfoma linfoblástico, linfoma folicular, linfoma de la zona marginal (linfoma tipo MALT-), mielodisplasia, mielofibrosis idiopática, trombocitemia esencial y algunas leucemias (leucemia megacarioblástica aguda, leucemia mielomonocítica crónica o mielodisplasia.33 En la mayoría de estos casos, el SH comparte características moleculares y citogenéticas con las neoplasias asociadas.33 Feldamn y cols han sugerido que el SH puede originarse por tres vías. Una, a partir de la transformación maligna de macrófagos tisulares maduros (SH genuino/primario), otra como resultado de transdiferenciación de otra neoplasia con diferente estirpe celular y la tercera, a partir de una neoplasia monocítica como la leucemia monocítica.34 Los siete casos informados por Fedman que presentaban SH secundarios a linfomas foliculares, mostraron la translocación t (14;18).33 Hay estudios recientes donde se han descrito casos raros de SH que se originaron de la transformación de linfoma folicular, fenómeno que recibe el nombre de transdiferencación.34,35 La transdiferenciación corresponde a la conversión de una célula progenitora madura a otra célula madura de linaje distinto. En el sistema hematopoyético existe un grado de plasticidad, especialmente entre las células dendríticas/histiocíticas y las células B. Experimentalmente se han determinado que linfocitos B pueden mostrar transdiferenciación hacia macrófagos, y a su vez diferenciarse hacia una célula precursora y subsecuentemente hacia células T, debido a la ganancia o pérdida de factores de transcripción como C/EBP, PAX-5.35 Igualmente, las células mieloides pueden diferenciarse hacia células dendríticas en presencia de interferón alfa.36 Es asombroso saber qué células maduras, puedan ser capaces de tal transformación. ¿Cuál es la forma en que esto se controla y qué grado de plasticidad tienen las células diferenciadas?, ¿Las células se reprograman directamente hacia un tipo celular diferente (transdiferenciación) o sufren regresión hacia un estado inmaduro (desdiferenciación) y posteriormente se diferencian hacia otra células?37 Cobaleda y colaboradores han mostrado que puede haber desdiferenciación, durante la transformación de un linfocito B maduro hacia una células T funcional, y señalaron que la deleción del Pax5, es un factor importante en la diferenciación de células B y responsable de esta desdiferencación.37 De la misma forma, Xie y colaboradores han elegantemente expuesto, que por efecto de la expresión de factores de transcripción como el C/EBPα y C/EBPβ en linfocitos B diferenciados, estos rápida y eficientemente son reprogramados hacia macrófagos, mediante la inhibición del Pax5, que a su vez desregula (downregulation) el CD19 y estimula la expresión el factor de transcripicón PU.1 y el factor de la familia ETS, lo que condiciona la expresión (upregulation) del Mac-1 y otros marcadores mieloides.38
En diversas series, el SH afecta primordialmente sitios extraganglionares como el aparato digestivo, la piel y el sistema nervioso central. Sin embrago, como expusimos anteriormente, también puede primariamente afectar ganglios linfáticos y bazo.39 Existen informes de SH originados en sitios como la tiroides, cavidad nasal, pulmón, hueso, médula ósea y paladar,7,18,40,41 y hay casos informados de afección sistémica involucrando múltiples sitios, a lo que ha sido referido como "histiocitosis maligna".42
¿ HISTAPATOLOGÍA
La imagen histológica del SH puede ser variable, pero por lo general están compuestos de células grandes (> de 20 μm) usualmente de redondas a ovales, con pleomorfismo acentuado (Figura 4). Se han descrito células de aspecto epitelioide y fusiforme con formación focal de empalizadas, lo que puede simular tumores de vaina nerviosa periférica.43
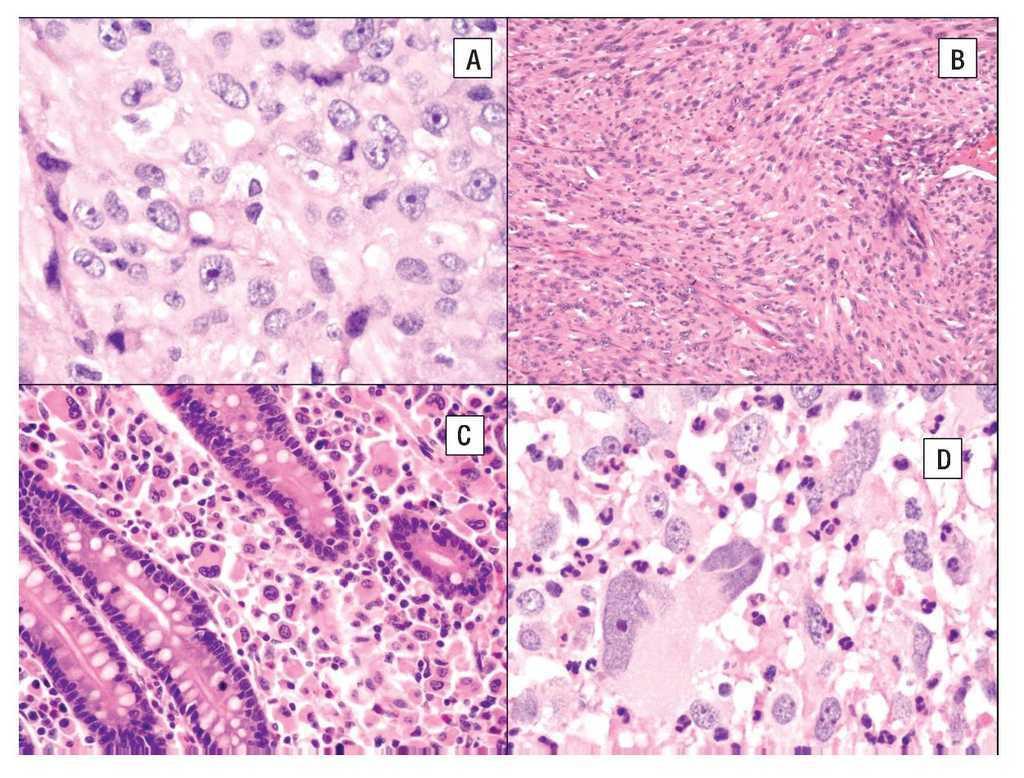
Figura 4. Histopatología del SH. A) Células grandes con abundante citoplasma eosinófilo (epitelioides) con pleomorfismo moderado. B) Aspecto fusiforme del sarcoma histioítico. C) Colon infiltrado por SH con células pleomórficas de aspecto epitelioide. D) Células de SH con pleomorfismo acentuado asociado a abundante infiltrado inflamatorio de neutrófilos.
El citoplasma es abundante y eosinófilo y pueden tener vacuolas finas. Es usual encontrar células bi o multinucleadas (Figuras 5A y 5B) y ocasionalmente hemofagocitosis por las células neoplásicas (Figuras 5C y 5D). Los núcleos generalmente son redondos a ovales o irregularmente lobulados, y a menudo son excéntricos con cromatina usualmente vesicular. Las figuras mitóticas varían de uno a 64 por 10 campos de x40,7 con áreas variables de necrosis y angio-invasión. Un dato que es muy característico de estas neoplasias, es la presencia de células reactivas asociadas como linfocitos, células plasmáticas, histiocitos y eosinófilos en variable cantidad, que en ocasiones pueden ser tan abundantes que las células neoplásicas aparecen ocultas por el intenso infiltrado inflamatorio, lo simula un proceso reactivo. Esta última característica es particularmente común en el SH que involucra sistema nervioso central.39 Cuando el SH afecta ganglios linfáticos, generalmente pierde su arquitectura por la proliferación de células neoplásicas citológicamente malignas que semejan histiocitos (Figura 5D).
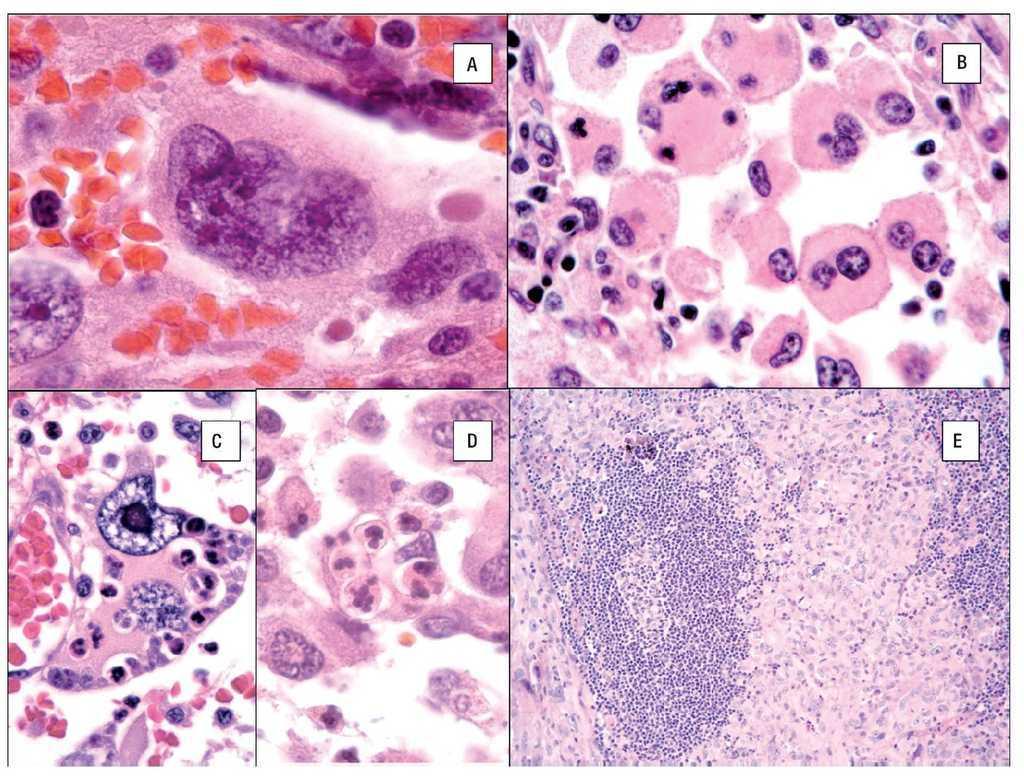
Figura 5. Histopatología del SH. Células bi o multinucleadas A y B) y hemofagocitosis por las células neoplásicas C y D). Macrófagos neoplásticos (histiocitos) rodeando un centro germinal. E).
Los estudios de microscopía electrónica han demostrado que las células neoplásicas presentan abundante citoplasma con numerosos lisosomas y ausencia de gránulos de Birbeck, complejos de unión y tono o microfilamentos (43), lo que las diferencia del sarcoma de células de Langerhans y del sarcoma de células dendríticas folicular (Figura 6).
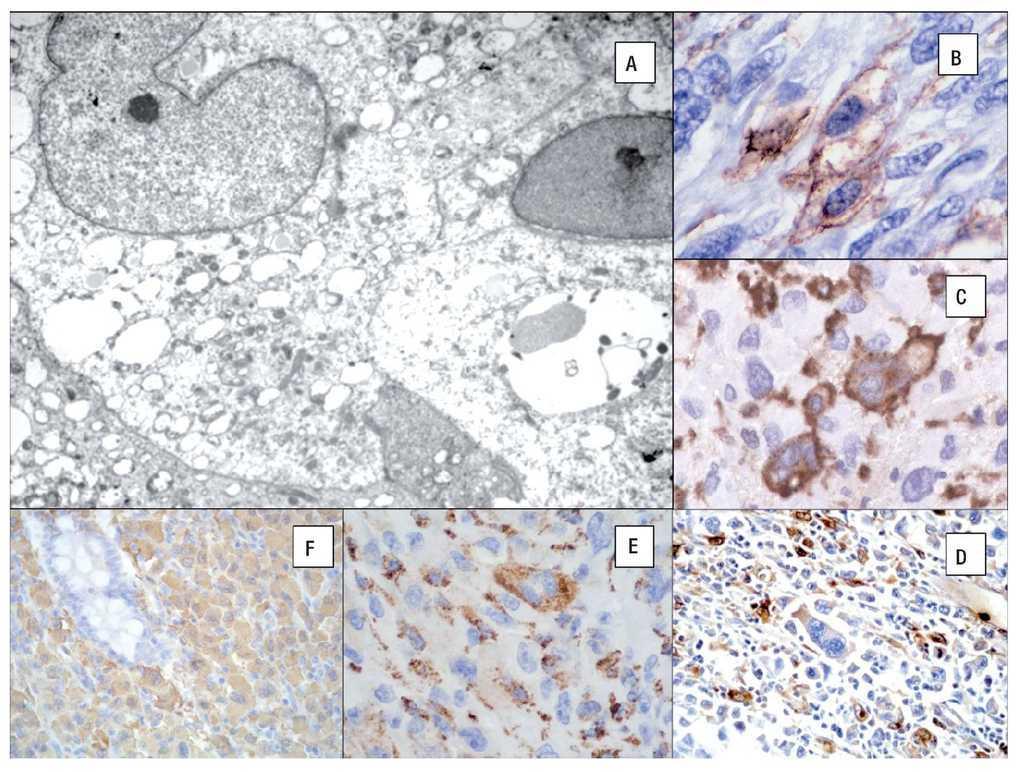
Figura 6. Microscopía electrónica de un caso de sarcoma histiocítico que muestra abundantes lisosomas. A). Inmunohistoquímica del SH: CD45 B), CD163 C), Lisozima D), CD68 (PGM-1) E), Fascina F). (Cortesía Dr. Hugo Domínguez-Malagón, Instituto Nacional de Cancerología México DF).
¿ INMUNOHISTOQUÍMICA
La inmunomarcación es fundamental para distinguir el SH de otras neoplasias de células grandes como el linfoma, melanoma o carcinoma.44 Por definición, el SH debe expresar CD45 además de dos o más marcadores histiocíticos que incluyen el CD163, CD68 (KP1 y PGM1), CD14 y lisozima (muramidasa) con ausencia de CD1a, Langerina, CD21, CD35, CD33, CD13 y mieloperoxidasa, además deberán ser negativos a citoqueratinas y marcadores melanocíticos (Figura 6). Otros anticuerpos para macrófagos (histiocíticos) que pueden ser utilizados son el HAM 56, a1antitripsina y la a1antiquimiotrisina. La fascina también puede ser positiva en el SH, pero no es un anticuerpo específico. Esta es una proteína (54-58 kD), originalmente asilada de los erizos de mar y asociada a la actina formadora de fascículos (de ahí el nombre; fascin de latín bundle), que regula el citoesqueleto celular. Es positivo en diversos tumores que pueden confundirse histológicamente con el SH como el Xantogranuloma juvenil (100%), el sarcoma sinovial (100%), sarcoma de células dendríticas interdigitantes (94%), el sarcoma de células dendríticas foliculares (93%), histiocitoma fibroso maligno (78%) y el fibrosarcoma (14%).45
Algunos otros marcadores que no son primariamente monocítico/macrofágicos, pero pueden emplearse en este contexto son el CD4, el CD45RO, el CD38 y HLA-DR.46 El CD4 es una molécula que interactúa con el HLA clase II, durante el reconocimiento antigénico y está presente en los linfocitos T CD4. Se ha encontrado también expresado en monocitos, células de Langerhans y algunas otras células dendríticas.18 Algunos tumores han sido positivos al marcador vascular CD31, que no es de extrañar puesto que el CD31 es positivo también en macrófagos normales.47
Los marcadores para macrófagos más utilizados son el CD68 (KP-1 y PGM-1), la lisozima, el CD 14 y el CD163. Tanto el CD68 (KP1 y PGM1) como la lisozima muestran expresión citoplásmica granular, además la lisozima muestra acentuación en la región del aparato Golgi (punto paranuclear). El CD 14 es un excelente marcador de monocitos/macrófagos y fue el primer "receptor de reconocimiento de patrones" descrito (PRC/ Pattern Recognition Receptor), que corresponde a un grupo de proteínas expresadas por células del sistema inmunológico nativo, que sirven para identificar moléculas asociadas a diversos microorganismos. El CD14 se expresa primordialmente en macrófagos (con marcación citoplásmica granular difusa) y débilmente en neutrófilos y algunas células dendríticas. Recientemente, se describió la utilidad del CD163 como uno de los mejores marcadores para macrófagos y neoplasias derivadas de estos.48 El CD163, es una glucoproteína de 130-kD que pertenece a la superfamilia de receptores de cisteína (scavenger receptor cysteine-rich superfamily), cuya expresión es altamente específica para células de linaje monocito/ macrófago y marca tanto el citoplasma como la membrana.48 Al ser comparado con el CD68 (Kp1 y PGM1), el CD163 ha dado mejores resultados, por lo es considerado el anticuerpo de elección en el diagnóstico de SH.3,48
Es importante tener en mente, que el melanoma puede resultar positivo a muchos de los anticuerpos anteriormente mencionados. Por ejemplo, en un estudio realizado por Pernick y colaboradores en 1999, demostraron que los melanomas pueden tener inmunorreactividad en 91% a la a1antitripsina, 95% al CD68, 86% al HAM56, 26% al Mac387 y 7% a la lisozima.49 Raramente puede haber expresión de CD15 y de proteína S-100, cuando ocurre generalmente es débil y focal.44 Por definición, el SH no debe expresar marcadores de células B o T. Los SH son negativos a los marcadores de melanoma (HMB-45 y Melan-A) y epiteliales (EMA y citoqueratinas). La expresión del Ki-67 es muy variable.
Es de interés hacer notar, que hay casos publicados de SH que presentan expresión de proteína S-100 (además de la coexpresión del CD163, CD68 y CD45), que Miettinen ha llamado tumores híbridos.44,50,51
Recientemente se describió la expresión de las proteínas de mucina- inmunoglobulinas de células T (TIM) 3 y 4, en macrófagos y células dendríticas de tejidos humanos:52 el más específico de estos, dos es el TIM4, porque el TIM-3 se ha encontrado en algunos linfocitos T TH1 y células T citotóxicas.53,54 Estas proteínas pertenecen a una familia de receptores fosfatidilserina localizados en la superficie de células T, y son importantes en el reconocimiento celular y la fagocitosis de células apoptóticas. Dorfman y colaboradores analizaron con inmunomarcación con anti-TIM 3 y anti-TIM 4, un grupo de neoplasias de estirpe histiocítico/dendrítico donde incluyeron al xantogranuloma juvenil, histiocitosis de células de Langerhans, el sarcoma de células de Langerhans, la neoplasia blástica de células dendríticas plasmocitoides (tumor hematodérmico), el sarcoma histiocítico, el sarcoma de células dendríticas foliculares y el sarcoma de células dendríticas interdigitantes.16 El resultado fue que todas ellas mostraron positividad en la totalidad de los casos estudiados, excepto el sarcoma de células dendríticas folicular. Esta falta de expresión en el sarcoma de células dendríticas foliculares tiene un cuestionamiento interesante, puesto que apoya la hipótesis de que esta neoplasia deriva de una célula madre mesenquimatosa, más que de una célula madre mieloide.55 Diversos carcinomas poco diferenciados, el melanoma y los linfomas de células grandes, resultaron negativos para el TIM3 y TIM4. El empleo de estos anticuerpos es muy prometedor, pues reduciría en gran medida la lista de neoplasias incluidas dentro del diagnóstico diferencial.56
¿ GENÉTICA
La patogénesis molecular del SH es incierta. Existe evidencia genética de una interacción de genes supresores tumorales PTEN (phospahatase and tensin homologe) y p16 (INK4A)/p14 (ARF). Estos genes han mostrado inactivación epigenética o genética en SH de ratones y de humanos, constituyendo un modelo sistémico para la patogénesis del SH.57
¿ DIAGNÓSTICO DIFERENCIAL
Las neoplasias incluidas en el diagnóstico diferencial incluyen:
Melanoma: El melanoma es uno de los grandes simuladores por su diversidad morfológica. Si bien es cierto, que al identificar pseudoinclusiones nucleares, nucléolos y citoplasma abundante eosinófilo, sugiere el diagnóstico de melanoma, se requiere corroborar por inmunohistoquímica la expresión de marcadores melanocíticos como la proteína S-100, HMB-45 y Melan A. Se debe tener precaución al interpretar la proteína S-100, ya que algunos casos de SH presentan positividad aunque débil y focal.3
Carcinoma metastásico: El pleomorfismo y la hemofagoctosis (aunque rara) presente en algunos carcinomas poco diferenciados, puede simular un SH. Sin embargo, la cohesividad de las células, la positividad a diversas citoqueratinas y al antígeno epitelial de membrana (EMA), presente en los carcinomas, ayudan a aclarar el diagnóstico.
Tumor/sarcoma de células dendríticas interdigitantes: Estas neoplasias generalmente afectan ganglios linfáticos, pero pueden presentarse en sitos extraganglionares como la nasofaringe, bazo, pleura, aparato digestivo, retroperitoneo, parótida, pulmón, amígdalas palatinas y testículo.44 En ganglios linfáticos, la lesión puede en un inicio, ser paracortical (sitio normal de localización normal de las células dendríticas interdigitantes) con patrón estoriforme y células fusiformes, que semejan un sarcoma fusocelular con poco pleomorfismo. Aunque las células son predominantemente fusiformes, pueden también ser redondas y ovaladas. Los núcleos son ovoides y característicamente presentan nucléolos pequeños. Por inmunohistoquímica, hay positividad difusa e intensa para proteína S-100, fascina y vimentina y negatividad a CD1a, Langerina, CD21, CD35 y CD23 y CD163. Algunos casos pueden expresar de forma débil y focal el CD68, lo cual no debe alarmar al patólogo.
Histiocitosis/sarcoma de células de Langerhans: En la histiocitosis de células de Langerhans típica (previamente llamada Histiocitosis X), las células proliferantes son fácilmente reconocibles. Estas son células grandes de núcleos contorneados y hendiduras, nucléolos pequeños y abundante citoplasma eosinófilo. Generalmente están acompañadas de un número variable de eosinófilos. Los casos que muestran macada atípia (sarcoma de células de Langerhans), suelen ser difíciles de distinguir de SH, sin embargo la expresión de CD1a, proteína S-100 y Langerina proporcionan una herramienta objetiva para el diagnóstico de estas neoplasias. El análisis ultraestructural demuestra la presencia de gránulos de Birbeck, que son estructuras citoplásmicas en forma de bastón o "raquetas", cuya función es inducida por la Langerina, y son características de las células de Langerhans. En los tumores de células de Langerhans puede haber expresión variable de CD68 y negatividad a la lisozima, CD21 y CD35.
Sarcoma de células dendríticas folicular (SCDF): Morfológicamente el SCDF es muy similar al sarcoma de células dendríticas interdigitantes, pues ambos presentan proliferación de células fusiformes y patrón estoriforme con bordes celulares poco definidos. Por inmunomarcación el SCDF se caracteriza por la expresión de dos o más marcadores de células dendríticas foliculares, como el CD21,CD35 y CD23 y la negatividad para S-100, CD1a, langerina actina, desmina y queratina.58 Puede haber expresión también de fascina, clusterina, Glut-1 y Claudina 1.59,60 Por microscopia electrónica, las células presentan procesos citoplásmicos largos interdigitantes con uniones celulares tipo desmosomas, con ausencia de gránulos de Birbeck.
Linfoma Anaplásico de células grandes (LACG): Descrito por Stein y cols en 1985, afecta principalmente ganglios linfáticos, se caracteriza por proliferación pleomórfica de células grandes con afección primordial a los sinudoides, que puede extenderse hacia la zona paracortical. Las células neoplásicas pueden presentar núcleos grandes de contorno irregular, por lo que se pueden confundir con un SH. La inmunomarcación es necesaria, y las células del LACG expresan diversos marcadores T (aunque hasta 20% de los casos pueden ser nulos), CD30 en la membrana, citoplasma y con acentuación en la zona del aparato de Golgi. Además, hay positividad al CD45, TIA-1, granzima B, perforina y ALK-1 hasta en el 70% de los casos, que es el resultado de la translocación t(2;5) (p23;q35). En la clasificación de la OMS 2008, se incorpora una variante de linfoma difuso de células grandes B, que expresa ALK-1. Esta variante presenta proliferación de células grandes, con pleomorfismo variable, con la presencia de nucléolos prominentes y morfología plasmocitoide. Estas células al ser negativas al CD20 y CD3, pueden orientar a que se trate de SH. Sin embrago, pueden expresar otros marcadores B como el CD79a, CD19 y Pax5 pero son negativas al CD68, CD163, CD14, lisozima y CD4. Las células del LACG B, además son positivas al CD30, EMA y ALK-1, este último con patrón granular citoplásmico.
Xantogranuloma Juvenil (XGJ): La familia del XGJ tiene características que comparte con la histiocitosis de células de Langerhans. Generalmente se presentan como tumores en piel, pero puede haber afección sistémica (hígado, bazo, pulmón, meninges y ojo).26 Morfológicamente hay proliferación de numerosas células con características clásicas de macrófagos (histiocitos), células gigantes tipo Touton (85%) y gran cantidad de histiocitos xantomatosos (espumosos). Por inmunohistoquímica hay expresión de marcadores histiocíticos como el CD163, CD14, CD68 (KP-1 y PGM-1) y son negativos al CD1a, Factor XIIIa, y a la proteína S-100, aunque si aparece suele ser débil y focal hasta en 20% de los caso.26
¿ PRONÓSTICO Y FACTORES PREDICTIVOS
El tamaño del tumor y el estadio de la enfermedad suelen tener valor pronóstico, mientras que la mitosis y el índice de proliferación celular medido con el Ki-67suelen ser muy variables y no ha demostrado tener relación directa con el curso clínico.40,61 Los SH son neoplasias agresivas con respuesta pobre al tratamiento, aunque hay informes en donde los pacientes con enfermedad clínica localizada y pequeños tumores primarios, tuvieron un curso clínico más favorable.7 La mayoría de los pacientes (60- 80%) se presentan en estadios clínicos avanzados (estadio II o IV), y mueren por progresión de la enfermedad. Algunos estudios, han determinado que el grado de la neoplasia y los márgenes quirúrgicos libres, son importantes para el pronóstico.43
¿ CONCLUSIONES
A pesar de ser una neoplasia poco frecuente, el SH debe ser considerado en el diagnóstico diferencial de sarcomas no sólo con morfología epitelioide y pleomórfica, sino también aquellos que presentan de células fusiformes.62 Generalmente hay infiltrado inflamatorio asociado, que en ocasiones puede ser muy intenso. Esta neoplasia puede semejar otras neoplasias linfoproliferativas, tanto por la presentación clínica como por la apariencia histológica. Y a pesar de la inmunomarcación, el diagnóstico puede no ser sospechado si no se incluyen, dentro de la batería de inmunohistoquímica, marcadores histiocíticos. Por inmunohistoquímica, se deben de excluir linfomas de células grandes, como los linfomas anaplásicos de células grandes, melanomas, carcinomas metastásicos poco diferenciados y algunos otros sarcomas. A pesar de que los SH son tumores agresivos, algunos casos con presentación clínica localizada, pueden tener curso clínico favorable.7
Correspondencia: Dr. Carlos Ortiz-Hidalgo.
Departamento de Patología, Centro Médico ABC. Sur 136 N° 116, Colonia Las Américas, CP 01120. México DF.
Teléfono y fax: (55) 5230 8171.
Correo electrónico:cortiz@abchospital.com