







Introducción: El Dermatofibrosarcoma Protuberans (DFSP) es una neoplasia rara. Se origina en la dermis, es de lento crecimiento, de aspecto nodular o polipoide que invade la hipodermis y por lo general no da metástasis. Los objetivos de este estudio fueron investigar la frecuencia, características clínicas y patológicas del DFSP en nuestro Hospital.
Material y métodos: Se buscaron en los archivos de Patología del Hospital de Oncología (IMSS), todos los casos de DFSP que ocurrieron en el periodo 2004 a 2008. Todos los casos fueron revisados y evaluados, de acuerdo con los criterios actuales para esta neoplasia. La información clínica se obtuvo de los expediente de cada paciente
Resultados: Noventa casos de DFSP se estudiaron, 56 (62%) casos tenían el aspecto típico de DFSP, mientras que en 10 (11.11%) casos, el DFSP mostraba áreas de fibrosarcoma. El tumor se localizó con mayor frecuencia en el tronco. La edad de los pacientes varió de 16 a 83 años en el momento del diagnóstico inicial. La relación hombre:mujer fue de 1:1. La mayoría de los casos se presentaron como placas o pequeños nódulos.
Conclusiones: El DFSP representa el 4% de todos los sarcomas de partes blandas en nuestro Hospital, las características clínicas y patológicas fueron similares a las reportadas en la literatura médica.
Introduction: Dermatofibrosarcoma Protruberans (DFSP) is a rare tumor originating from the dermis, it is slow growing, nodular, polypoid neoplasm that invade the subcutaneous tissue. Distant metastasis occurs with extreme rarity.
This study aimed to identify frequency, clinical and pathologic factor associated with DFSP in our Hospital
Material and methods: The archives of the Department of Pathology, Mexican Oncology Hospital (IMSS) were retrospectively searched to identify cases of DFSP diagnosed from 2004 to 2008. All cases were reviewed and evaluated using current criteria. Patient´s clinical information was obtained from the medical records.
Results: Ninety cases of DFSP were studied. The histological findings were typical of DFSP in 56 (62%) cases, whereas in 10 (11.11%) cases, areas with a fascicular or "herringbone" growth pattern, considered to represent fibrosarcomatous change. Tumor location were similar in both groups, with the trunk being the most common site. The patients ranged from 16 to 83 years at the time of initial surgical excision. The male:female ratio was 1:1. Most of the lesions began as plaque-like areas or small raised nodules.
Conclusions: DFSP constitutes 4% of all soft tissue sarcomas in our Hospital. Clinical and pathologic features were similar to those described in the literature.
INTRODUCCIÓN
El Dermatofibrosarcoma Protuberans (DFSP) es un tumor mesenquimatoso cutáneo de crecimiento lento, localmente agresivo, con un alto índice de recurrencia después de la escisión quirúrgica, sin embargo, rara vez da metástasis.1 Puede afectar cualquier grupo de edad, con mayor incidencia en la cuarta década de la vida. El sitio anatómico más afectado es el tronco, seguido por las extremidades, la cabeza y el cuello.2
Se desconoce su histogénesis, actualmente se considera que se origina en una célula madre mesenquimal.3 Se caracteriza por una translocación lineal entre los brazos largos de los cromosomas 17 y 22 t(17;22)(q22;q13), o por un cromosoma supernumerario en anillo derivado de ésta misma translocación,4 lo cual permite la amplificación del factor b de crecimiento derivado de plaquetas, que juega un papel importante en la patogénesis del DFSP. Esta mutación hace que el DFSP sea susceptible a terapia con inhibidores de quinasas.
Es un tumor raro, representa 0.1% de todos los tumores malignos y 1.8% de los sarcomas de tejidos blandos.5 En Estados Unidos, se ha calculado que se presentan de 0.8 a 4.5 casos por millón de habitantes al año. Afecta todas las razas, siendo dos veces más frecuente en población negra que en la caucásica.6
El objetivo del presente trabajo es establecer la frecuencia del DFSP y sus variantes morfológicas en un Hospital de Oncología, analizar sus características clínicas y los problemas más comunes en el diagnóstico diferencial.
MATERIAL Y MÉTODOS
Se recolectaron de los archivos del Servicio de Patología todos los casos diagnosticados como "Dermatofibrosarcoma Protuberans", del primero de enero de 2004 al 31 de diciembre del 2008. Fueron revisados de segunda intención por dos patólogas y una dermatopatóloga. En la evaluación de los casos, se utilizaron los criterios morfológicos previamente establecidos.7 En los casos con dificultad diagnóstica se emplearon marcadores de inmunohistoquímica como: CD34, HMB45, Melan A, proteína S100 y actina de músculo liso. Los datos clínicos se obtuvieron de los expedientes de cada paciente. Los resultados se expresaron mediante estadística descriptiva, las variables sociodemográficas en base a medidas de tendencia central en caso de ser cuantitativas, y de distribución normal si eran cualitativas.
RESULTADOS
CARACTERÍSTICAS CLÍNICAS
Se obtuvieron 110 casos diagnosticados como DFSP, sin embargo al revisarlos por segunda intención, se corroboró el diagnóstico de DFSP en 90 casos. En el mismo periodo evaluado, se estudiaron 2 200 casos de sarcomas de tejidos blandos, por lo cual la frecuencia de los casos de DFSP fue del 4%, con respecto al número total de los mismos. Cuarenta y cuatro se presentaron en mujeres y 46 en hombres, con una relación hombre:mujer de 1:1.
La localización más frecuente fue la región del tronco con 51% de casos, seguido por la cabeza y cuello (24.3%) y en tercer lugar extremidades inferiores (13%). Dos casos (2.2%) se presentaron en la vulva.
El rango de edad de los pacientes fue de 16 a 83 años, con un promedio de 43 años de edad, en ambos géneros. Los adultos de 31 a 50 años fueron los más afectados por esta neoplasia, representando 41% de los casos.
En la mayoría de los pacientes (74%), la lesión inició con una pápula pequeña indurada, de color rojo, de lento crecimiento, que posteriormente se tornó en una placa atrófica o esclerótica. En 11% de los pacientes, la lesión también inició como un nódulo indurado para luego transformarse en un tumor multinodular subcutáneo. El 15% de los pacientes acudieron a consulta cuando la neoplasia formaba un nódulo central ulcerado, de aproximadamente 4 cm de diámetro mayor, bordeado por placas adyacentes induradas, en donde la piel adquirió un color violáceo.
El tiempo de evolución de la enfermedad fue variable, con un periodo de dos a 20 años (mediana cinco años). Por otro lado, dos pacientes de 58 y 62 años de edad respectivamente, señalaron que dicho nódulo apareció cuando tenían alrededor de 25 y 30 años, respectivamente.
Catorce (16%) de los pacientes tenían una historia de cuatro a seis resecciones previas, antes de ser admitidos en el Hospital de Oncología.
CARACTERÍSTICAS ANATOMOPATOLÓGICAS
Aspecto macroscópico. El tamaño de los especímenes varió de 4 a 19 cm (mediana: 6 cm). En términos generales, las lesiones < 6 cm tenían un aspecto de placa, con la epidermis intacta, por otro lado, el espesor medio de la piel y tejido celular subcutáneo al corte fueron de 4.5 cm. Las lesiones > 10 cm tenían un aspecto nodular, con bordes poco definidos, ulceraban la epidermis suprayacente y la dermis adyacente tenía un espesor de 3 a 4 cm. En todos los casos, la superficie de corte fue blanco-grisácea, fibrosa, pero en tres casos era completamente mixoide.
Aspecto microscópico. La mayoría de los casos (62%) estaba compuesto por células fusiformes, dispuestas en un patrón en "rueda de carreta" (Figura 1), con o sin un centro colagenoso acelular central. El número de mitosis promedio en 10 campos a seco fuerte fue de uno a cuatro.
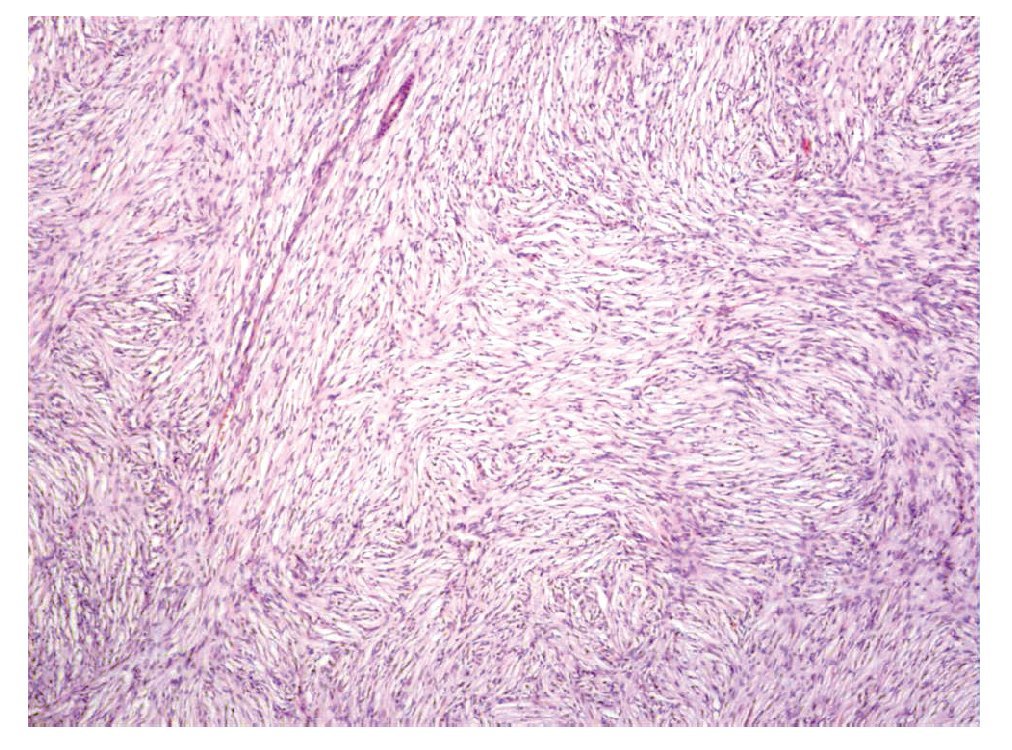
Figura 1. Dermatofibrosarcoma Protuberans variedad convencional, neoplasia fusocelular con patrón en haces cortos que se entrecruzan formando rehiletes (Hematoxilina & eosina).
En tres (4.3%) de los casos, la neoplasia mostró áreas extensas mixoides (Figura 2), poco celulares, con escasos vasos, ovales, que mostraron el aspecto clásico del DFSP en el 10% al 20% de su superficie. Tres casos mostraron depósito de melanina (Figura 3), y dos tenían áreas con estructuras similares a los cuerpos de Verocay (Figura 4). Por otro lado, cinco (7.1%) casos mostraron áreas extensas de esclerosis.

Figura 2. Dermatofibrosarcoma Protuberans con áreas mixoides.

Figura 3. Dermatofibrosarcoma Protuberans pigmentado, constituido por células fusiformes y epitelioides, con gránulos de melanina en su citoplasma (Hematoxilina & eosina).

Figura 4. Corte histológico que señala la formación de cuerpos de Verocay en un Dermatofibrosarcoma Protuberans.
En diez (11.11%) de los casos, la neoplasia mostró una transición entre áreas clásicas de un DFSP y áreas compuestas por una neoplasia mesenquimatosa hipercelular, con fascículos largos que adoptaban un patrón de crecimiento en "espina de pescado" (Figura 5). El número de mitosis en estas áreas en 10 campos a seco fuerte fue de ocho a 15. Estos casos se diagnosticaron como DFSP con diferenciación a fibrosarcoma.

Figura 5.Corte histológico en donde se ilustra la transición abrupta entre Dermatofibrosarcoma Protuberans convencional y fibrosarcoma.
El diagnóstico de DFSP fue modificado en 20 casos, ocho casos fueron reclasificados como Histiocitoma Fibroso Profundo, tres como Fibromatosis Músculo-Aponeurótica, tres como Sarcoma de Kaposi, cuatro como Angiosarcomas y dos como Leiomiosarcoma.
En 12 casos (13%), el diagnóstico de DFSP fue difícil de establecer, ya que la neoplasia mostraba características poco convencionales que nos obligaron a descartar otras opciones de diagnóstico:
En tres casos se consideró la posibilidad diagnóstica de dermatofibroma celular, ya que las lesiones no mostraron áreas francas de invasión en el tejido celular subcutáneo, sin embargo, la positividad intensa al CD34, favoreció el diagnóstico de DFSP.
En dos casos, la neoplasia mostró áreas pleomórficas, por lo que se planteó también la posibilidad diagnóstica de Sarcoma Dérmico de alto grado, sin embargo, debido a que el 90% de la neoplasia mostró el aspecto clásico del DFSP e intensa positividad al CD34, el diagnóstico final en tales casos fue de DFSP con focos de diferenciación a sarcoma de alto grado.
En tres casos más, la epidermis estaba por completo ulcerada, la neoplasia mostró un patrón de crecimiento fusocelular con pigmento en algunas células, la consideración diagnóstica más importante fue la de un melanoma, sin embargo, la positividad intensa al CD34 y la negatividad a marcadores de inmunohistoquímica tales como el HMB45 y el Melan A, descartaron el diagnóstico mencionado.
Finalmente, en los tres casos de DFSP con extensas áreas mixoides, se planteó también el diagnóstico de una neoplasia neurogénica, la negatividad a la PS100 y el aspecto característico del DSFP en otras áreas, apoyó el diagnóstico del mismo.
El DFSP en general presentó un índice mitósico bajo, menor a cinco mitosis en 10 campos a seco fuerte en 93% de los casos, no obstante, en los casos con transformación a fibrosarcoma (11.11%), el índice mitósico aumento con un promedio de seis a 10 mitosis en 10 campos a seco fuerte.
DISCUSIÓN
El DFSP es una neoplasia poco frecuente, representando en la literatura médica el 1.8% de todos los sarcomas de tejidos blandos.5 En nuestra serie su frecuencia fue del 4%. La edad promedio de presentación fue de 43 años, similar a lo reportado en la literatura médica.8
En cuanto a la topografía de la neoplasia, el sitio más afectado fue el tórax, seguido por la cabeza y el cuello, estos resultados coinciden con lo encontrado por varios autores en sus series de casos.9
Dos de los casos se presentaron en la vulva, lo cual representa una topografía poco común. En 1999, Ghorbani10 reportó cuatro ejemplos similares..
El aspecto clínico del DFSP está determinado en gran medida por el tiempo de evolución de la neoplasia. Los patrones clínicos iniciales del DFSP, se han denominado estadios preprotuberantes11 y, pueden manifestarse como: a) una placa indurada, blanca o marrón, con aspecto de cicatriz, b) una lesión semejante a atrofodermia, caracterizada por una placa de consistencia blanda y deprimida, blanca o marrón que recuerda atrofia de la piel o anetodermia y, c) similar a hemangioma, compuesta de placas rojas o violáceas, induradas o blandas, que recuerdan lesiones vasculares.
En nuestra serie, el 74% de los pacientes presentaron una pápula pequeña indurada, de color rojo, de lento crecimiento, que posteriormente se tornó en una placa atrófica o esclerótica. Sin embargo, el 11% de los pacientes presentaron inicialmente un nódulo indurado que se transformó en un tumor multinodular subcutáneo. Los pacientes con DFSP con transformación sarcomatosa, mostraron por lo general grandes masas, con bordes infiltrantes. Llombart y colaboradores12 en una serie de 20 casos, encontraron características macroscópicas similares a las reportadas en esta serie.
El aspecto microscópico característico del DFSP consiste en haces cortos de células fusiformes, que se disponen en un patrón de crecimiento denominado en "rueda de carreta".13,14 El 62% de nuestros casos mostró dichas características, por otro lado, la mayoría de los casos mostraron escasas mitosis, de uno a cuatro por 10 campos a seco fuerte. Estas características son similares a lo reportado por la mayoría de los autores, ya que los casos descritos en sus series, mostraron pleomorfismo leve y una actividad mitósica baja, que no excedió de cinco mitosis por 10 campos a seco fuerte.15
El DFSP puede mostrar diferentes subtipos histológicos o variantes tales como la variante mixoide,16,17 la esclerótica,18,19 la mioide,20 la pigmentada,21 con células granulares,22 con cuerpos de Verocay.23 Además de las variantes descritas, resulta importante tomar en cuenta que hay casos de DFSP que pueden transformarse en sarcomas de alto grado,23 fenómeno que ocurrió en 10 de nuestros casos.
En nuestra serie, 12 casos fueron de difícil diagnóstico debido a que mostraban características morfológicas diferentes a los DFSP convencionales. En estos casos, además del análisis morfológico, la evaluación de la expresión de marcadores de inmunohistoquímica como el CD34, HMB45, Melan A, Proteína S100, entre otros, nos ayudaron a realizar el diagnóstico correcto.
CONCLUSIONES
- El DFSP representa el 4% de todos los sarcomas de tejidos blandos en nuestro Hospital.
- Es importante reconocer las variantes morfológicas de esta neoplasia para evitar errores de diagnóstico.
- El empleo de marcadores de inmunohistoquímica como el CD34, Melan A, Actina de Músculo liso, Proteína S100, entre otros, son de gran ayuda en casos de difícil diagnóstico.
Correspondencia:
Dra. Isabel Alvarado Cabrero.
Departamento de Patología, Hospital de Oncología, Centro Médico Nacional Siglo XXI, IMSS.
Av. Cuauhtémoc #330, Colonia Doctores.
C.P. 06720. México D.F., México.
Teléfono: 5627 6900, extensión 22733.
Correo electrónico: keme2.tijax12@gmail.com