




INTRODUCCIÓN
En los últimos años ha ido apareciendo un número creciente de medicamentos genéricos cuyas indicaciones, recogidas en su ficha técnica, difieren de las que tiene el medicamento de referencia. Detrás de este hecho no hay una diferencia farmacológica o una falta de bioequivalencia entre ambos medicamentos, sino un conflicto de patentes de segunda indicación que impide reflejar todas las indicaciones del genérico temporalmente.
Para entender este caso es necesario abordar previamente la importancia de las diferentes patentes que pueden afectar a los medicamentos.
PATENTE Y PERÍODO DE EXCLUSIVIDAD: DIFERENTES TIPOS DE PROTECCIÓN
La patente de los medicamentos queda bajo el ámbito de la Ley 11/1986, de 20 de marzo, de Patentes de Invención y Modelos de utilidad (BOE nº 73, 26-Mar-1986), que adaptó la normativa comunitaria sobre patentes a nuestro ordenamiento jurídico. Según esta norma, en su artículo 49 se indica: "la patente tiene una duración de 20 años improrrogables, contados a partir de la fecha de presentación de la solicitud y produce sus efectos desde el día en que se publica la mención de que ha sido concedida". No olvidemos que lo que se patenta son moléculas y que deben demostrar si van a ser medicamentos algún día, primero en un laboratorio y después ensayando en seres humanos. Las exigencias de los expedientes de autorización implican unos tiempos mínimos de investigación en los que se invierten una media de 10 años (algunos más, como por ejemplo los medicamentos biológicos, y otros menos, como las combinaciones de principios activos conocidos). Por lo tanto, la explotación del "invento" se reduce y siempre ha sido objeto de queja de la industria farmacéutica. Para compensarlo, la Unión Europea creó el Certificado Complementario de Protección para los medicamentos que otorga hasta 5 años de ampliación de la patente para estos productos (previa solicitud)1. Para fomentar la investigación se ha ampliado la patente en 6 meses a los medicamentos que realicen estudios para demostrar la indicación y/o uso pediátrico2.
Aunque existen varios convenios internacionales sobre la patente (suscritos por España casi todos), éstos sirven para homogeneizar criterios pero no existe aún una patente común válida para todos los Estados, ni siquiera en la Unión Europea. Ello obliga al titular del medicamento a solicitar la patente en cada país donde quiera comercializar el producto. Esto no siempre se hace de manera simultánea, por lo que los 20 años no vencerán al mismo tiempo.
Entre los tipos de patente que se pueden otorgar a los medicamentos están los siguientes:
Patente de procedimiento
Este tipo de patente protege una serie de operaciones mediante las cuales se transforman unos compuestos iniciales en uno o varios productos finales. Viene regulado en el artículo 50 de la ley 11/19861 y sería el caso de una síntesis orgánica que conduce a un medicamento X a partir de una serie de materias primas y productos intermedios. Este tipo de patente promovió una amplia difusión de medicamentos "copia" en numerosos países, entre ellos España. Antes de la entrada en vigor de la ley, era muy común la comercialización de productos que son copias de productos originales, pero obtenidos por procedimientos diferentes al del laboratorio original. Es preciso recordar que la diferencia de un medicamento "copia" con un genérico es que este último ha demostrado su intercambiabilidad o bioequivalencia.
Patente de producto
La patente de producto farmacéutico representa la máxima protección que se puede obtener para una invención, consistente en un nuevo compuesto químico que se ha sintetizado en un laboratorio de investigación. Un compuesto nuevo puede ser patentado siempre y cuando tenga una aplicación industrial. De lo contrario, nos hallaríamos en presencia de un mero descubrimiento.
En España, esta patente se reconoció tras la entrada de nuestro país en la Unión Europea (entonces CEE) en 1986, aunque no se aplicó hasta 1992, cuando finalizaron los periodos transitorios de la Ley 11/19861. Las negociaciones para la adhesión de España a la CEE tuvieron una intensa negociación en este sentido, dada la fuerte oposición de la industria farmacéutica nacional a la inclusión de la patente de producto, al comprender que era la que otorgaba al inventor la mayor protección; ello estaba motivado por el volumen de medicamentos "copia" existentes en nuestro mercado. La simple protección de los productos farmacéuticos mediante patentes de procedimiento se había revelado claramente insuficiente, pues aquel que inventa un procedimiento nuevo para la obtención de idéntico producto podía explotarlo libremente sin necesidad de la autorización del titular de la primera patente.
Patente de segunda indicación
De acuerdo con el criterio del Convenio de Munich sobre la Concesión de Patentes Europeas3 ratificado por España, las reivindicaciones de segundas indicaciones son adecuadas para proteger nuevos usos de medicamentos ya conocidos, si la nueva indicación se dirige a sectores específicos de la población (tales como pacientes con una susceptibilidad médica concreta o personas con un genotipo determinado). También, una segunda indicación puede proceder de una modalidad de administración diferente del mismo medicamento (administración subcutánea y administración intramuscular), siempre y cuando el método de administración sea nuevo y contenga actividad inventiva.
Este tipo de patente generó controversia en sus orígenes al ser interpretado de diferente manera entre los países que ratificaron el Convenio de Munich (finales de los ochenta). Sobre todo, existían dudas de interpretación sobre si lo que realmente se autorizaba era la patente de un tratamiento para el cuerpo humano, algo prohibido en la mayoría de las legislaciones nacionales (véase el artículo 4.6 de la Ley 11/19861; sin embargo, la primera indicación terapéutica conocida de un compuesto químico conocido es patentable a pesar de esta exclusión). Tras una modificación de la redacción del mismo, se acordó otorgar la patente de segunda indicación bajo la siguiente fórmula: "El uso de (sustancia conocida) para la fabricación de un medicamento para el tratamiento de (una enfermedad concreta)". Por este motivo, el titular de una patente de segunda indicación estará legitimado para impedir a un competidor la comercialización, ofrecimiento, utilización o promoción del compuesto para el nuevo uso protegido pero, sin embargo, nunca podrá prohibir que los médicos prescriban el uso para la segunda indicación de un producto que ya está en el mercado para la primera indicación4.
Periodo de exclusividad de datos
Por su parte, la autoridad sanitaria, aunque no tiene competencias en regulación de la propiedad industrial, no es ajena a los problemas potenciales que afecten a la industria farmacéutica, la cual, por otro lado, es un sector muy importante desde el punto de vista económico. De este modo, la normativa que regula los medicamentos se ve jalonada de detalles interpretables como una protección y como ayuda al fomento de la investigación y desarrollo (I + D) de la industria. Con ello se pretende generar un marco legal que a la vez que exige medicamentos seguros, eficaces, de calidad con información e identificación adecuada y un balance beneficio/riesgo favorable, mantenga la rentabilidad para los titulares de medicamentos innovadores.
Una de las medidas de protección más relevantes es el denominado "periodo de exclusividad" mediante el cual, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) impedirá la comercialización de medicamentos genéricos hasta transcurridos diez años desde la fecha de la autorización inicial del medicamento de referencia. Este período de diez años se ampliará hasta un máximo de once años si, durante los primeros ocho años del período de diez años, el titular de la autorización de comercialización del medicamento de referencia obtiene una autorización para una o varias indicaciones terapéuticas nuevas y, durante la evaluación científica previa a su autorización, se establece que dichas indicaciones aportarán un beneficio clínico significativo en comparación con las terapias existentes5. Este periodo es independiente de las patentes.
En función del tiempo empleado en la investigación, el periodo de exclusividad que concede la autoridad sanitaria puede ser mayor que el de la patente (fig. 1). De ahí la importancia que tiene para los titulares de medicamentos nuevos. En cualquier caso son distintos. La violación de la patente origina una demanda ante los juzgados de lo mercantil que produce como medida cautelar la inmovilización inmediata del producto demandado hasta que se resuelva el contencioso; esto está ocurriendo con cierta frecuencia en los últimos años, como en el caso de los genéricos de atorvastatina que se intentaron comercializar a finales de 2008 o recientemente con escitalopram Sandoz® EFG.
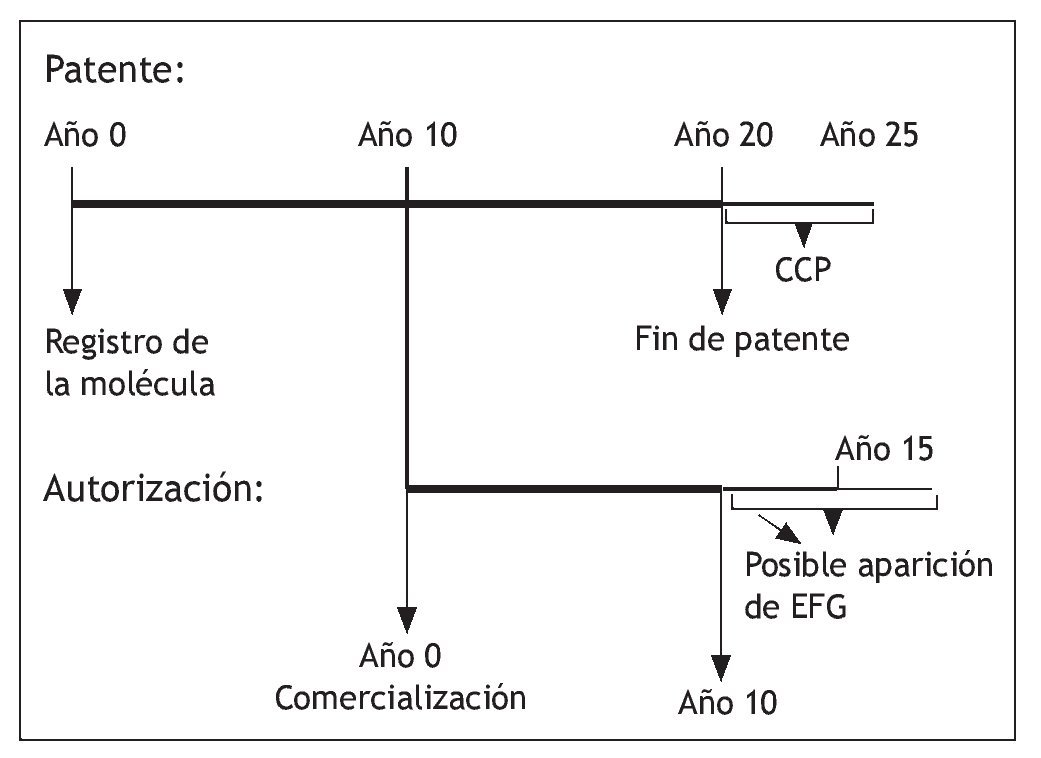
Fig. 1. Patente y autorización de medicamentos. CCP: certificado complementario de patente.
LAS INDICACIONES DE LOS GENÉRICOS Y SUS MEDICAMENTOS DE REFERENCIA
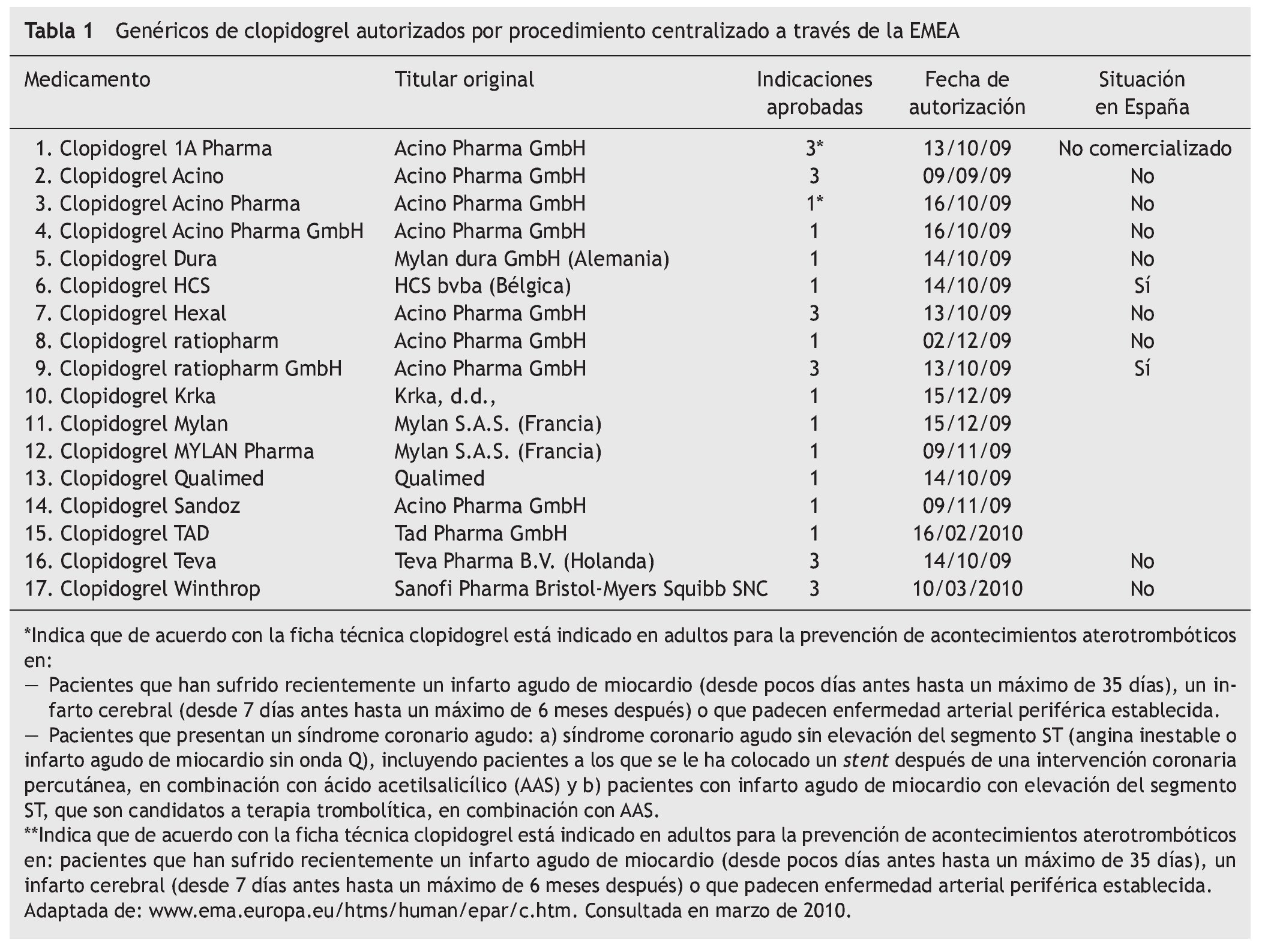
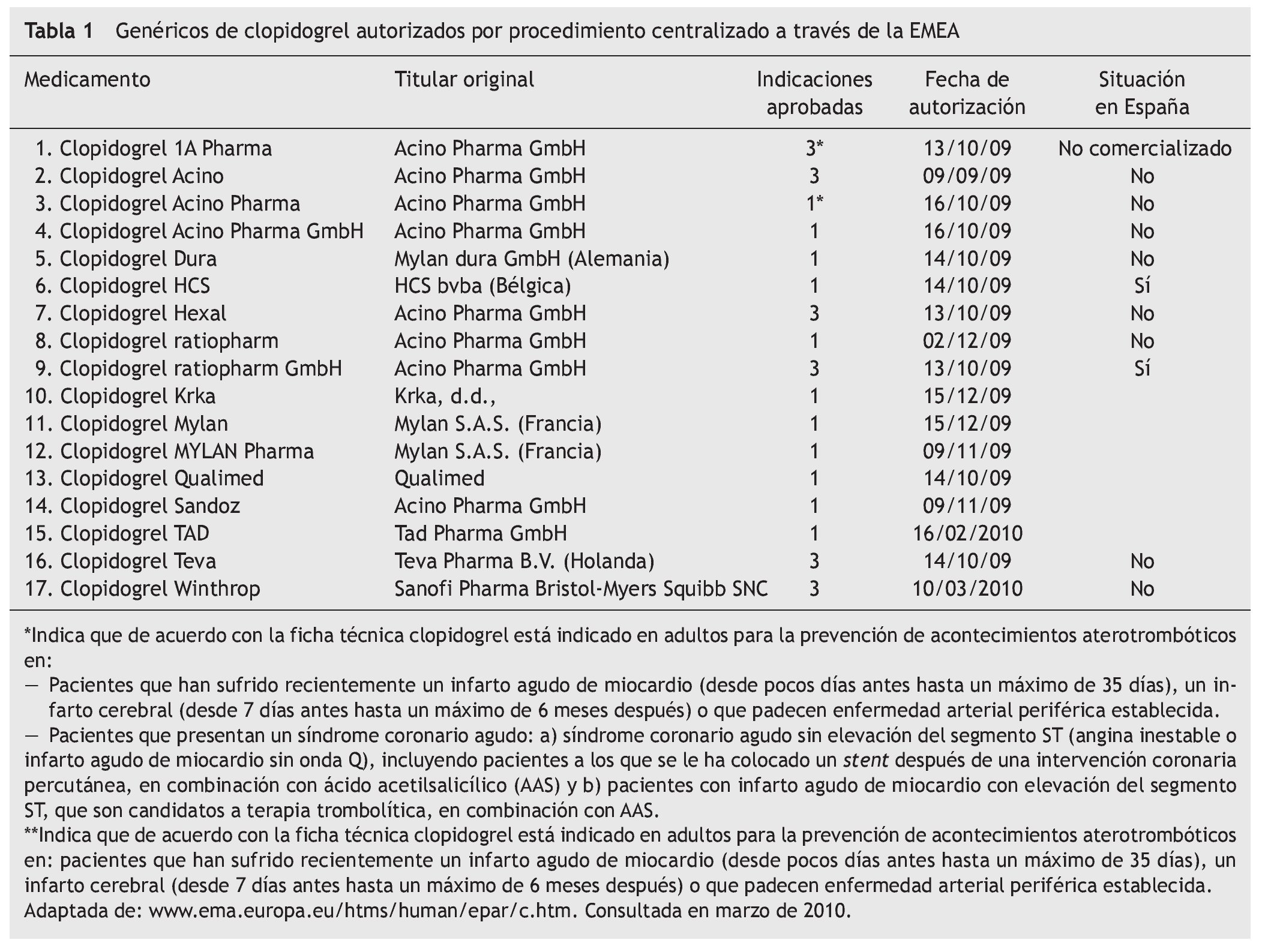
Tras la aplicación de la patente de segunda indicación, se viene observando con frecuencia la aparición en el mercado de medicamentos genéricos que no contienen todas las indicaciones que el medicamento de referencia. Como ejemplo podemos citar la paroxetina, cuya indicación para el "trastorno de estrés postraumático" no está recogida en todas las fichas técnicas de medicamentos genéricos con este principio activo. Más recientes son los casos de diferentes medicamentos genéricos del clopidogrel. También es inminente la comercialización del escitralopram genérico, la mayoría de ellos recoge solamente una de las cinco indicaciones del medicamento de referencia (Cipralex®) el "tratamiento de episodios depresivos mayores".
Esta situación puede desconcertar, sobre todo en el caso de los principios activos que con la misma composición cualitativa y cuantitativa han ido patentando diferentes indicaciones.
En este sentido, algunos titulares de medicamentos han optado por diferenciar, a través de marcas comerciales, lo que es indistinguible desde un punto de vista químico. Un ejemplo de ello son los fármacos que contienen ropinirol 0,25 mg (Requip® y Adartrel®). El primero se comercializó en 1997 para el tratamiento de la enfermedad de Parkinson y el segundo en 2006 para el síndrome de las piernas inquietas. Ambos tienen una idéntica composición cualitativa y cuantitativa. El genérico de Adartrel® no debería aparecer antes del año 2016 (debido al periodo de exclusividad de datos otorgado por la autoridad sanitaria, sin perjuicio de la patente), mientras que los genéricos de Requip® ya están en el mercado. En ambos casos, el genérico estará vinculado a las indicaciones de su medicamento de referencia. Es preciso constatar que, ante una receta de Adartrel®, si el farmacéutico dispensador se encuentra ante uno de los supuestos legítimos para la sustitución de medicamentos previstos en la ley, no existe ningún impedimento para sustituirlo por un ropinirol EFG, respetando siempre los requisitos de la sustitución.
¿Por qué lo permite la normativa?
De acuerdo con el artículo 37 del Real Decreto 1345/2007, se permite (con intención de excepcionalidad) que la ficha técnica de un genérico no contemple las mismas indicaciones que un medicamento de referencia, si alguna de ellas está amparada por la patente o el periodo de exclusividad de datos en su caso. Esto parece contradecir el principio de bioequivalencia e intecambiabilidad entre medicamentos de referencia y genéricos, al menos en lo que a información a profesionales se refiere. Pero detrás de ello hay una solicitud del titular del medicamento genérico para que la autoridad reguladora, la AEMPS o la Agencia Europea del Medicamento (EMEA) lo permitan con carácter excepcional.
Este aspecto hay que dejarlo claro y no se debe permitir que se utilice para fomentar una hipotética diferencia entre la actividad farmacológica de un principio activo presentado en diferentes medicamentos: clopidogrel genérico, a una concentración determinada, tendrá todos los efectos adversos e indicaciones recogidas en la ficha técnica del medicamento original. Resulta absurdo pensar que un determinado principio activo va a diferenciar su actividad farmacológica en función del nombre del envase que lo contiene, habiendo demostrado su intercambiabilidad.
A la autoridad sanitaria no le interesa permitir la comercialización de un genérico, sabiendo que al día siguiente puede quedar inmovilizado y suspendido cautelarmente por orden de un juzgado de lo mercantil. Ello tendría como consecuencia que no se pueden aplicar los precios de referencia que tanto ahorro nos van a generar, como ya ha ocurrido con los genéricos de olanzapina, entre otros, en 20086.
Para facilitar la labor al médico prescriptor, el legislador puso a su disposición el artículo 85 de la Ley 29/2006: "Las administraciones sanitarias fomentarán la prescripción de los medicamentos identificados por su principio activo en la receta médica. En los casos en los que el prescriptor indique en la receta simplemente un principio activo, el farmacéutico dispensará el medicamento que tenga menor precio y, en caso de igualdad de precio, el genérico, si lo hubiere". No se habla de indicaciones y es perfectamente legal la dispensación del medicamento genérico ante una receta con prescripción por principio activo para cualquiera de sus indicaciones.
La Ley 29/2006 también apoya este aspecto en su artículo 93.4, a la hora de la sustitución, indicando: "Cuando se prescriba un medicamento que forme parte de un conjunto y que tenga un precio superior al de referencia, el farmacéutico deberá sustituirlo por el de menor precio e idéntica composición cualitativa y cuantitativa en principios activos, forma farmacéutica, vía de administración, dosificación y presentación que el medicamento prescrito y, en caso de igualdad de precio, por el medicamento genérico". Esto se mantendrá así, a menos que se trate de principios activos no sustituibles por el farmacéutico, descritos en la normativa7. En este caso, tampoco se vincula la sustitución a la indicación, como era de esperar.
En el caso de clopidogrel, medicamento de diagnóstico hospitalario sometido a visado en la dispensación, puede generar la duda adicional de si será denegada la dispensación de un genérico al carecer de las indicaciones del original. Cabe recordar que el objetivo del visado es verificar la conformidad del tratamiento prescrito en el Sistema Nacional de Salud con las condiciones de utilización autorizadas en la ficha técnica y las indicaciones terapéuticas financiadas de acuerdo con el procedimiento que determinen en el ejercicio de sus competencias las CC. AA"8. Resulta necesario dar a conocer a los órganos autonómicos encargados del visado el motivo por el que las fichas técnicas no tienen las mismas indicaciones, tal como se ha descrito anteriormente.
Tanto el legislador europeo como el nacional entienden que una patente de segunda indicación no tiene por qué otorgar una protección adicional para las patentes de la primera indicación ya vencidas. Por ese motivo, nuestra legislación permite la salida al mercado del genérico sin todas las indicaciones. Esto es un fiel reflejo de la compleja situación legal actual generada por una normativa, tanto estatal como europea, que intenta buscar un equilibrio entre la industria farmacéutica "innovadora" y la de "medicamentos esencialmente similares". La Administración y sus gobiernos necesitan encarecidamente a las dos. Detrás de un nuevo medicamento normalmente existe una intensa labor investigadora que dinamiza un sector clave para las economías de cualquier país. Es indudable que promueve una potente actividad económica y empresarial que genera muchos puestos de trabajo: ningún gobierno quiere renunciar a ello. El problema es que esta I + D genera un producto que va a estar financiado en la mayoría de los casos, y la factura la pagan las administraciones públicas: ningún gobierno quiere renunciar a reducir este gasto. Los medicamentos genéricos ponen en bandeja un ahorro tan necesario que llega a su tiempo, cuando la patente ha caducado y cuando el medicamento original ha tenido tiempo suficiente para demostrar su valía: está amortizado y no se daña la I + D.
También conviene recordar los ingresos económicos que suponen para las agencias reguladoras la llegada de nuevas solicitudes de autorización de medicamentos. No importa que existan más de 60 "omeprazoles" en el mercado; sean bienvenidos todos los que quieran, siempre que cumplan con los requisitos que marca la ley. A través de la tabla 1 se puede hacer un cálculo de las tasas que cobra la Administración (AEMPS en este caso) por cada solicitud relacionada con los medicamentos. Además de las tasas por evaluación de medicamentos, existen otras tasas de carácter periódico como la tasa de intención de comercializar o la tasa de evaluación de informe periódico de seguridad de un medicamento (de acuerdo con la exigencias del Sistema Español de Farmacovigilancia), por la que pasan todos los medicamentos recogidos en el Nomenclator. Hagan sus cuentas y verán que la comercialización de nuevos medicamentos, genéricos incluidos, también es un buen negocio para la Administración.
¿Por qué hay genéricos que incluyen todas las indicaciones?
La aparición de los medicamentos genéricos ha obligado a adaptar las estrategias comerciales de las principales multinacionales farmacéuticas. Una de ellas consiste en la creación de empresas filiales que comercializan genéricos, en concreto genéricos de sus propios medicamentos, fabricados por el mismo que fabrica el original pero bajo una marca distinta y un titular diferente (su filial); ejemplos: Sanofi® tiene a Winthrop®; Novartis® tiene a Sandoz®, Tedec-Meiji® tiene a Mabo®, etc. Como es de esperar, no va a tener ningún problema de patente y en su ficha técnica se van a recoger todas las indicaciones del producto original. Se trata del mismo producto, al cual se le cambia el etiquetado primario y secundario para poner las siglas EFG. Obligatoriamente debe salir al mercado al precio de referencia o inferior, si lo desea, y compite directamente con los demás genéricos. Está tan claro que es el mismo producto que la normativa le exime de demostrar que es intercambiable5. Algunos los denominan "autogenéricos".
Dado que la mayoría de los Servicios de Salud fomentan la prescripción por principio activo o de genéricos, con esta estrategia consiguen acceder a la porción de mercado de sus competidores directos. Se ha detectado que en algunos casos se publicitan como "el único genérico idéntico al medicamento de referencia" o "el único genérico con todas las indicaciones que el original"; son mensajes de legalidad cuestionable dado que el lector puede interpretar una diferencia en eficacia y/o calidad que no existe.
Un ejemplo de ello es el Clopidogrel Ratiopharm GmbH® 75 mg. El titular del medicamento es Acino Pharma GmbH®, empresa dedicada a desarrollos farmacéuticos con sedes en Suiza y Alemania, que a su vez comercializa su propio clopidogrel genérico en varios países de Europa y que permite a Ratiopharm GmbH® su comercialización (en forma de medicamento licenciatario genérico, ver artículo 7 y 12 del Real Decreto 1345/2007). Se autorizó por procedimiento centralizado a través de la EMEA en octubre de 2009. Los motivos por los que puede contener todas las indicaciones son:
— Dispone de permiso del titular del medicamento original, en este caso es Sanofi Pharma®-Bristol-Myers Squibb SNC®. Esto es poco probable pero no imposible.
— El titular del medicamento original no puede impedirlo por caducidad de la patente de segunda indicación.
— Al titular del medicamento original no le interesa litigar en este asunto.
El autor de este artículo preguntó por este asunto a los responsables de Ratiopharm en España y señalaron que desde su matriz en Alemania les habían confirmado la inexistencia de conflicto de patentes. El tiempo nos dirá si QRV GLUi VL están en lo cierto o si su medicamento queda inmovilizado tras la denuncia en el juzgado de los titulares de Plavix® e Iscover®. Si no ha ocurrido ya, está claro que no hay conflicto.
Tras esta situación, están ciertas maniobras comerciales habituales entre los medicamentos de todo tipo, como son las licencias de comercialización. Al igual que empresas Fcomo Pfizer® conceden licencias a terceros para comercializar sus productos (véase el caso de atorvastatina, donde Cardyl® se comercializa también como Zarator® y Prevencor®, marcas de otras empresas licenciatarias), con los titulares de medicamentos genéricos ocurre lo mismo; cuando consiguen demostrar la bioequivalencia que va a permitir su autorización, es común que se llegue a acuerdos comerciales similares como el protagonizado entre Acino Pharma GmbH® y Ratiopharm GmbH® con clopidogrel (tabla 2). El primero permite al segundo la utilización del expediente de autorización para poner en el mercado "su" clopidogrel.

En el caso del clopidogrel genérico, la empresa Acino Pharma GmbH® hizo varias solicitudes distintas, basadas en medicamentos diferentes pero con idéntico principio activo (tabla 1). La única diferencia entre las fichas técnicas de ambos productos son las indicaciones. Parece que los solicitantes no tenían claro los resultados de un posible conflicto de patentes de segunda indicación a su favor y apostaron por lo seguro: solicitar autorización para el mismo medicamento bajo marcas distintas y con información de indicaciones en ficha técnica diferente (véanse filas 2, 3 y 4 de la tabla 1 y filas 8 y 9). Una vez despejadas las dudas sobre la posibilidad de comercializar un genérico de clopidogrel con todas las indicaciones, éste es el elegido para la comercialización. De los dos genéricos de clopidogrel de Ratiopharm GmbH®, sólo comercializan en España el que más les interesa.
Aunque se han hecho esfuerzos por aumentar la transparencia e información en los expedientes de evaluación de medicamentos, queda de manifiesto una importante laguna que puede ser fácilmente subsanable. Sería muy útil que los profesionales sanitarios pudieran consultar en la propia ficha técnica o en alguna base de datos de la AEMPS si la ausencia de indicaciones iguales entre dos medicamentos con la misma composición cualitativa y cuantitativa obedece a motivos de patentes o a eficacia y/o seguridad no intercambiable.
Esto se hace especialmente necesario cuando se ha publicado recientemente una norma como el Real Decreto 1015/2009, de 19 de junio, por el que se regula la disponibilidad de medicamentos en situaciones especiales. Debe quedar claro cuándo el médico prescriptor está ante una situación de utilización de medicamentos autorizados en condiciones diferentes a las establecidas en su ficha técnica, tal como se describe en el capítulo III de la norma. Esta norma estaba diseñada para dar cobertura legal a la prescripción puntual de, por ejemplo, levodopa en el síndrome de piernas inquietas, no recogido en su ficha técnica al no haberse demostrado. Si bien la norma ha eliminado burocracia al médico prescriptor de acuerdo con su artículo 13, esta práctica "tendrá carácter excepcional y se limitará a las situaciones en las que se carezca de alternativas terapéuticas autorizadas para un determinado paciente, respetando en su caso las restricciones que se hayan establecido ligadas a la prescripción y/o dispensación del medicamento y el protocolo terapéutico asistencial del centro sanitario. El médico responsable del tratamiento deberá justificar convenientemente en la historia clínica la necesidad del uso del medicamento e informar al paciente de los posibles beneficios y los riesgos potenciales, obteniendo su consentimiento conforme a la Ley 41/2002, de 14 de noviembre".
Debe quedar claro que esta situación no tiene nada que ver con la petición excepcional de no incluir las indicaciones amparadas bajo una patente de segunda indicación realizada por el titular de un medicamento genérico a la autoridad evaluadora correspondiente.
LA MARCA EN LOS MEDICAMENTOS GENÉRICOS
Otro caso que demanda explicaciones es la aparición de genéricos con nombre de fantasía. En particular, resulta curioso el caso de Neobrufen® 600 mg EFG. Se trata de un "autogenérico" cuyo titular es Abbott Laboratorios S.A., que en este caso ha preferido mantener la marca del original. Como se ha explicado anteriormente, lo normal es que no deba aportar datos preclínicos ni clínicos para demostrar la equivalencia con el medicamento de referencia por ser el mismo medicamento que el de referencia.
¿Para qué sacar al mercado un genérico de su propio producto? Exclusivamente por interés comercial. La intervención administrativa de los precios de los medicamentos obliga a adaptarse al precio de referencia cuando aparece el genérico (si no lo hace es como si quedara fuera de la prestación farmacéutica del Sistema Nacional de Salud). Al lanzar su "autogenérico" se consigue más presencia en el mercado.
No obstante, cabe resaltar que la intención de la normativa es que tras los medicamentos de referencia las "copias" autorizadas, cuando corresponda, sean medicamentos genéricos, ya que éstos han demostrado su intercambiabilidad. No se concederán las siglas EFG a medicamentos que no se ajusten a la definición de medicamentos genéricos o cuando la bioequivalencia no pueda ser demostrada por medio de estudios de biodisponibilidad. Por ese motivo, en abril de 2007 la solicitud de autorización de Neobrufen® 600 mg fue revocada por la AEMPS para luego ser autorizado como Neobrufen® 600 mg EFG en agosto de 2008. Se supone que es bioequivalente a Brufen® 600 mg comprimidos recubiertos, siendo el mismo medicamento con la única diferencia del acondicionamiento final.
La denominación de los genéricos en España se puede hacer de dos formas5:
— Con la denominación oficial española del principio activo y, en su defecto, con la denominación común usual o científica de dicha sustancia acompañada, en su caso, del nombre o marca del titular o fabricante.
— Con una marca, siempre que no pueda confundirse conuna denominación oficial española o una denominación común internacional ni inducir a error sobre las propiedades terapéuticas o la naturaleza del medicamento.
Este aspecto ya se contemplaba en el apartado IV de la exposición de motivos de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, tal como se solicitaba a España desde la Comisión de la UE, puesto que en otros países comunitarios los genéricos podían tener nombre de fantasía desde hacía años.
Tenemos en el mercado varios casos de este tipo: como diferentes genéricos que tienen como principio activo quetiapina: Psicotric®, Qudix®, Ilufren®, etc. Sabemos que son genéricos porque deben llevar las siglas EFG. Esto se contempló en la normativa ante las reivindicaciones de fabricantes de genéricos, asociaciones de consumidores y pacientes por varios motivos:
— Para facilitar la identificación de los medicamentos y que los pacientes tengan mayor facilidad para nombrarlos en lugar de hacerlo a través de su principio activo.
— Para mejorar la imagen de los medicamentos genéricos y asimilarlos más a los de referencia.
— Para facilitar su prescripción por parte de algunos profesionales médicos que sienten animadversión a todo lo que "huela" a genérico.
CONCLUSIONES
1. La ficha técnica de un medicamento genérico puede no tener las mismas indicaciones que el medicamento de referencia por motivos de patente de segunda indicación, no por diferencias en su composición, eficacia o seguridad.
2. Para un medicamento dado, no se podrá prohibir que los médicos prescriban el uso de un genérico del mismo para cualquiera de sus indicaciones, siempre que ésta requiera la misma dosificación del mismo principio activo. El genérico ha demostrado su intercambiabilidad.
3. Se necesita mayor transparencia para que la información de la ficha técnica indique si la ausencia de indicaciones se debe a cuestiones de patentes de segunda indicación.
Correspondencia: José Manuel Paredero Domínguez.
Servicio de Farmacia. Gerencia de Atención Primaria de Guadalupe. C/ Ferial, 31, 3º. 19071 Guadalajara.
E-mail: malloret@sescam.org
Recibido el 30 de abril de 2010;
aceptado el 4 de junio de 2010