







Solid organ transplantation (SOT) is the best treatment option for end-stage organ disease. The number of SOT procedures has been steadily increasing worldwide during the past decades. This trend has been accompanied by the continuous incorporation of new antimicrobial drugs and by the refinement of strategies aimed at minimizing the risk of opportunistic infection. Nonetheless, viral infections, which can occur at any stage of the post-transplant period, remain a clinical challenge that negatively impacts both patient and graft outcomes. This review offers an overview of the most relevant viral infections in the SOT population, with a focus on herpesviruses (cytomegalovirus, Epstein–Barr virus, varicella-zoster virus, and herpes simplex virus 1 and 2) and polyomaviruses (human BK polyomavirus). In addition, the currently recommended prophylactic and treatment approaches are summarized, as well as the new antiviral agents in different phases of clinical development.
El trasplante de órgano sólido (TOS) es la mejor opción para el tratamiento de la enfermedad orgánica terminal, con un incremento sostenido a lo largo de las últimas décadas. Esta tendencia ha estado acompañada por la constante incorporación de nuevos antimicrobianos y el perfeccionamiento de las estrategias destinadas a minimizar el riesgo de infección oportunista. No obstante, las infecciones virales, que pueden aparecer en cualquier momento de la evolución post-trasplante, siguen constituyendo un desafío clínico con un impacto negativo en la evolución del receptor como del injerto. La presente revisión ofrece una panorámica de las infecciones virales más relevantes en el receptor de TOS, con énfasis en los herpesvirus (citomegalovirus, virus de Epstein-Barr, virus varicela-zóster y virus herpes simplex 1 y 2) y poliomavirus (poliomavirus humano BK). Se repasan igualmente las estrategias de profilaxis y tratamiento actualmente recomendadas, así como los nuevos agentes antivirales en diversas fases de desarrollo clínico.
Solid organ transplantation (SOT) procedures have been steadily increasing over the past decades.1 SOT is generally considered a life-saving, cost-effective treatment for patients with end-stage organ disease.2,3 Nonetheless, this approach is not free from risk, and post-transplant complications can significantly contribute to patient's morbidity and mortality.4 Infections are one of the most common complications developing after transplantation.5 A relevant proportion of such events are produced by opportunistic microorganisms and remain ultimately associated to the long-term immunosuppressive therapy needed to avoid allograft rejection and loss.6 A number of guidelines have been developed to guide clinicians in the best prophylactic and preemptive strategies for SOT recipients according to their clinical characteristics and specific risk factors, thus contributing to reduce the incidence of overall and, specifically, opportunistic infection (OI).5,7,8 Nevertheless, viral infections—which can occur at virtually any stage of the post-transplantation follow-up—still constitute a major disease burden with a deleterious effect on both recipient and graft outcomes.9–11
The present review is focused on the most important viral pathogens in the SOT population according to their respective incidence and clinical impact: cytomegalovirus (CMV), Epstein–Barr virus (EBV), varicella-zoster virus (VZV), herpes simplex viruses (HSV) and human BK polyomavirus (BKPyV). We will summarize current recommendations on prophylaxis and treatment, as well as ongoing research concerning investigational antiviral agents and novel prevention strategies that are still in different phases of clinical development.
CMVCMV belongs to the Betaherpesvirinae subfamily within the Herpesviridae family.12 Seroprevalence (as assessed by the presence of CMV-specific immunoglobulin G [IgG] antibodies) in the general population reaches over 60% in developed countries and up to 100% in developing countries.13 In most cases, primary infection occurs at early ages and is asymptomatic. Nevertheless, CMV may cause severe clinical manifestations in susceptible subjects, such as seronegative pregnant women who seroconvert during pregnancy and suffer intrauterine infection with structural damage of the fetus, or patients with impaired cell-mediated immunity such as allogeneic hematopoietic stem cell transplant (allo-HSCT) recipients and those diagnosed with acquired immune deficiency syndrome (AIDS).13 Of note, SOT recipients are particularly susceptible to CMV infection, either in form of reactivation of a latent infection facilitated by post-transplant immunosuppression, or in form of primary infection or superinfection due to donor-derived transmission.
Clinical manifestations and risk factorsIt has been long described that CMV can exert the so-called “direct” and “indirect” effects in SOT recipients.14,15 The direct effects attributable to CMV refer to the clinical symptoms that result from viral replication, cytopathic effect and organ spreading.16 CMV disease is commonly categorized as viral syndrome (a febrile illness accompanied in most cases by laboratory abnormalities such as leucopenia or neutropenia, atypical lymphocytes, thrombocytopenia or elevation of liver aminotransferases) and end-organ disease, in which viral tissue invasion may manifest as colitis, hepatitis, pneumonitis, esophagitis and, more rarely, retinitis or myocarditis.16,17 The indirect effects associated to CMV replication (even at low levels) would be related in turn to the viral capacity for evasion of host responses and immunomodulation, leading to a proinflammatory cytokine milieu and modulation of the nitric oxide synthase pathway, which result in endothelial cell damage and accelerated vasculopathy.16,18 These CMV-associated indirect effects have been linked to a plethora of complications, including opportunistic infection such as listeriosis or Pneumocystis jirovecii pneumonia,19,20 invasive fungal disease,21,22 atherothrombotic events,23 vanishing bile duct syndrome after liver transplantation (LT),24,25 chronic graft dysfunction26 and, ultimately, graft loss.
Several risk factors for developing CMV infection and/or disease after SOT have been described. Recipients at the highest risk are those seronegative that receive an organ from a seropositive donor (D+/R− group), since these patients lack pre-existing CMV-specific cell-mediated immunity.27 The incidence rates of CMV viremia and disease can be as high as 90% and 50–65%, respectively, at three months after transplantation in the absence of adequate antiviral prophylaxis.27 On the contrary, seronegative recipients from a seronegative donor (D−/R−) face the lowest risk of CMV disease, with incidence rates ranging from 1 to 2% at the first year.16 The rates for CMV viremia and diseases among seropositive recipients (R+) are of 40–60% and 5–6%, respectively, although the D+/R+ category appears to have a higher risk than D−/R+ patients due to risk of superinfection with a different donor-transmitted viral strain.27,28 Seropositive patients treated with lymphocyte-depleting antibodies (such as polyclonal antithymocyte globulins or monoclonal antibodies targeting CD52 [alemtuzumab] or CD3 [OKT3]) also show an increased susceptibility to CMV disease.29 Finally, specific SOT populations like lung transplant (LuT) and small bowel transplant (SBT) recipients suffer from an increased incidence as compared to kidney transplant (KT) or LT recipients due to the higher amount of lymphoid tissue contained within the transplanted graft.27 In contrast, the use as maintenance immunosuppression of a mammalian target of rapamycin (mTOR) inhibitor associated to reduced-exposure calcineurin inhibitors (tacrolimus or cyclosporine) has been shown to exert a protective effect.30,31
Prevention strategies for CMV infectionDue to the relevance of CMV infection in the SOT setting, several scientific societies have issued specific recommendations on the choice of one or other of the two alternative approaches, namely universal prophylaxis or preemptive therapy, available to prevent this complication. Two of the most recent documents have been published by a joint committee from the Spanish Society of Transplantation (SET), the Group for Study of Infection in Transplantation of the Spanish Society of Infectious Diseases and Clinical Microbiology (GESITRA-SEIMC) and the Spanish Network for Research in Infectious Diseases (REIPI) in 201632 and by the American Society of Transplantation (AST) Infectious Diseases Community of Practice in 2019.33
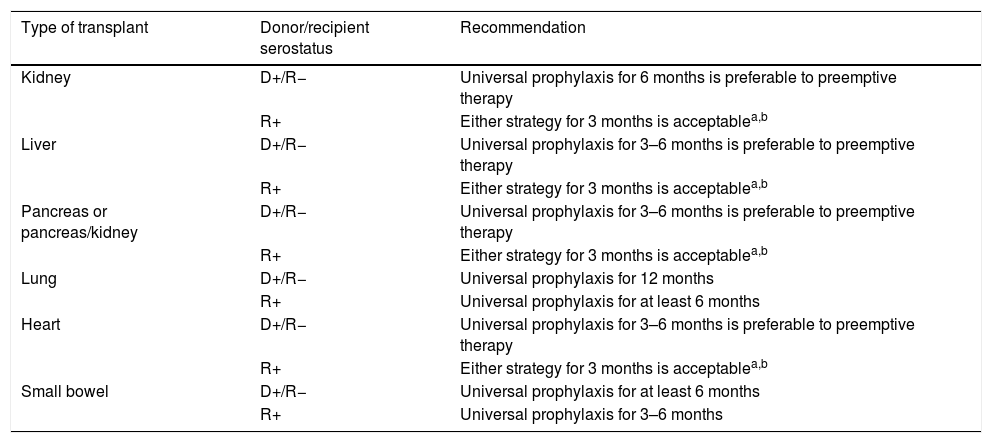
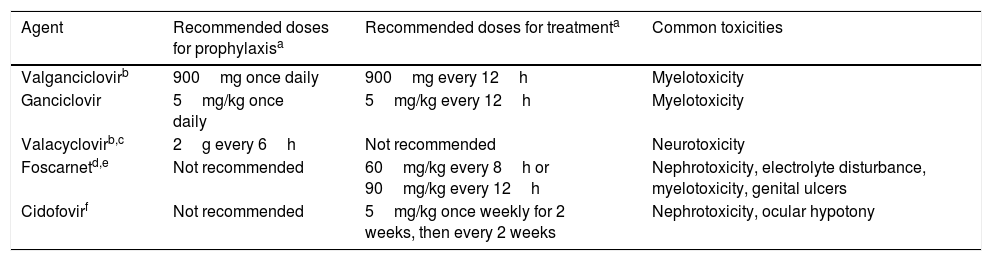
Prevention strategies for CMV infection will depend on the SOT population considered, the D/R serostatus constellation, and the presence of individual risk factors in the recipient (Table 1). Two major strategies can be used. Universal prophylaxis is based on the continuous administration of an antiviral drug with activity against CMV during the peak of post-transplant immunosuppression, whereas the preemptive therapy requires the monitoring of CMV antigenemia or, preferably, CMV DNAemia according to a pre-established schedule and the initiation of antiviral drugs once the viral load exceeds certain threshold in an otherwise asymptomatic recipient.32,33 In both scenarios, the preferred drugs are intravenous (IV) ganciclovir (GCV) and its oral L-valyl ester prodrug valganciclovir (VGCV) (Table 2).32,33 High-dose valacyclovir (2g every 6h) has been also used for the prevention of CMV infection after KT.34 Although the incidence rates of CMV disease and viremia in an open-label, single-center randomized trial were comparable to those observed with VGCV, KT recipients allocated to the valacyclovir arm experienced a significant higher incidence of biopsy-proven acute rejection35 and moderate-to-severe interstitial fibrosis and tubular atrophy.36 Universal prophylaxis is associated with an increased risk of drug-related adverse effects (namely neutropenia and leukopenia with VGCV) and drug-related costs, although offers logistical advantages and would presumably exert a protective impact on the indirect effects attributable to CMV.37 The preemptive approach, in turn, results in a lower potential for toxicity and reduced costs but is more difficult to coordinate, requires a rapid response from the microbiology department to provide physicians with the results of CMV viral loads within a short turnaround time, and still lacks standardized thresholds to dictate the requirement of treatment.33 Thus, it is recommended that each transplant center determines its own cut-off values,32 although it seems reasonable that D+/R− patients and those receiving lymphocyte-depleting agents could benefit from setting lower thresholds.38 The demonstration of increasing viral kinetics should also prompt the initiation of antiviral therapy.32
Recommendations for the prevention of CMV infection/disease in adult SOT recipients according to the type of transplant and D/R serostatus.
| Type of transplant | Donor/recipient serostatus | Recommendation |
|---|---|---|
| Kidney | D+/R− | Universal prophylaxis for 6 months is preferable to preemptive therapy |
| R+ | Either strategy for 3 months is acceptablea,b | |
| Liver | D+/R− | Universal prophylaxis for 3–6 months is preferable to preemptive therapy |
| R+ | Either strategy for 3 months is acceptablea,b | |
| Pancreas or pancreas/kidney | D+/R− | Universal prophylaxis for 3–6 months is preferable to preemptive therapy |
| R+ | Either strategy for 3 months is acceptablea,b | |
| Lung | D+/R− | Universal prophylaxis for 12 months |
| R+ | Universal prophylaxis for at least 6 months | |
| Heart | D+/R− | Universal prophylaxis for 3–6 months is preferable to preemptive therapy |
| R+ | Either strategy for 3 months is acceptablea,b | |
| Small bowel | D+/R− | Universal prophylaxis for at least 6 months |
| R+ | Universal prophylaxis for 3–6 months |
D: donor; R: recipient.
Antiviral drugs used for the prevention and treatment of CMV.
| Agent | Recommended doses for prophylaxisa | Recommended doses for treatmenta | Common toxicities |
|---|---|---|---|
| Valganciclovirb | 900mg once daily | 900mg every 12h | Myelotoxicity |
| Ganciclovir | 5mg/kg once daily | 5mg/kg every 12h | Myelotoxicity |
| Valacyclovirb,c | 2g every 6h | Not recommended | Neurotoxicity |
| Foscarnetd,e | Not recommended | 60mg/kg every 8h or 90mg/kg every 12h | Nephrotoxicity, electrolyte disturbance, myelotoxicity, genital ulcers |
| Cidofovirf | Not recommended | 5mg/kg once weekly for 2 weeks, then every 2 weeks | Nephrotoxicity, ocular hypotony |
Antigenemia testing—a semiquantitative assay that detects the lower matrix phosphoprotein pp65 (UL83) in CMV-infected peripheral blood leukocytes—and quantitative nucleic acid amplification testing (qNAAT) are the two most used methods for guiding preemptive prophylaxis and response to treatment.32,33 Antigenemia has been widely replaced by qNAAT due to the limitations of the former technique, such as the lack of standardization, its rather subjective interpretation, and the fact that it cannot be performed in patients with an absolute neutrophil count lower than 1000cells/μL. Therefore, real-time qNAAT is the currently recommended method for the monitoring of CMV infection.32,33 Nonetheless, qNAAT also has some limitations, such as the presence of notable analytical variability across laboratories according to the PCR platforms, clinical samples, gene target and nucleic acid extraction techniques used.39 Centers should decide on one assay and always use the same sample type (plasma or whole blood, but not both) whenever monitoring CMV DNAemia. On the other hand, assays must be calibrated according to the WHO International Reference Standard, and viral loads should be reported as international units (IU/mL) rather than copies.32,33
It is commonly assumed that high-risk groups benefit from universal prophylaxis. Therefore, a 6-month course of VGCV at prophylactic doses (900mg daily with adjustment for renal function) is recommended for D+/R− KT recipients, whereas shorter courses (3–6 months) should be used for LT, heart transplant (HT) and pancreas transplant (PT) recipients within this serological category.32,33 LuT recipients at risk must always receive VGCV prophylaxis for 6 (R+) to 12 months (D+/R−), although some centers extent prophylaxis beyond the first post-transplant year. Finally, SBT recipients also benefit from maintaining prophylaxis for a minimum of 6 months, although R+ recipients can receive only a 3-month regimen.33
Preemptive therapy is mainly reserved for KT, LT, HT and PT seropositive recipients (R+). In these populations, CMV viral loads must be ideally monitored at weekly intervals during the first 12 weeks after transplantation.33 Treatment (usually with oral VGCV) should be administered if the CMV viral load is above the threshold established as per institutional protocol. If the center does not have an adequate logistic support, a 3-month prophylaxis course is a valid alternative to the preemptive approach.
In the case of KT, LT, HT or PT seropositive recipients (R+) treated with lymphocyte-depleting agents as induction or anti-rejection therapy or within desensitization protocols, universal prophylaxis should be prescribed for 3–6 months (a 6-month course may be more effective than shorter regimens).32
The routine use of prophylaxis or preemptive therapy is not recommended among D−/R− recipients. Serological monitoring for CMV primary infection (i.e. IgM or IgG seroconversion) during the first year after transplantation may be useful if lymphocyte-depleting agents were prescribed or in the case of high-risk transplant groups (such as LuT, SBT and PT recipients).32 Moreover, D−/R− recipients should receive leukodepleted or CMV-seronegative blood products in order to avoid transfusion-related CMV infection. Although not routinely applied in all transplant centers, prophylaxis with acyclovir or valacyclovir could be considered during the early post-transplant period in order to prevent HSV-1 or HSV-2 reactivation in seropositive patients.33
Early post-transplant hypogammaglobulinemia (HGG), defined by a serum IgG level<600mg/dL at day 7, has been described as a risk factor for CMV disease in LuT40 and HT recipients.41 Intravenous CMV-specific hyperimmune globulin (CMV-HIG) or polyclonal intravenous immunoglobulins (IVIGs) associated with antiviral prophylaxis has been shown to reduce the incidence of CMV disease after LT,42 LuT43–45 and HT.44 Thus, some centers that perform LuT and HT have incorporated this adjunctive treatment to their clinical practice.33 Nonetheless, in the absence of adequate controlled trials, data supporting its efficacy are limited46,47 and debate persists about the indication of replacement therapy with CMV-IG or IVIG with the sole aim of preventing CMV disease, although high-risk patients with HGG in which no other preventive therapies can be implemented could likely benefit from this approach.32
Treatment of CMV infectionDue to the potentially severe consequences of CMV disease to both the recipient and the graft, antiviral treatment must be prescribed as soon as possible with oral VGCV and IV GCV as first-line options (Table 2). Parenteral therapy is preferred for life-threatening episodes, presence of high viral loads and in case of impaired gastrointestinal absorption.32,33 The duration of therapy must be individualized on the basis of the resolution of the clinical symptoms and demonstration of virological clearance, which should be assessed on a weekly schedule and always using the same laboratory test.33 Nevertheless, a minimum of two weeks of treatment is recommended.32,33
Secondary prophylaxis with VGCV for 1–3 months following the completion of treatment is not currently recommended as routine practice to avoid CMV recurrence due to the limited evidence of efficacy,48 but could be considered in selected patients (e.g. those with previous diagnosis of CMV invasive disease, augmented immunosuppression or LuT recipients).32,33
The development of CMV resistance to GCV must be suspected whenever CMV DNAemia and/or clinical symptoms persist or increase after a two-week course of therapy.38 Risk factors for GCV resistance include D+/R− serostatus, high viral loads at the initiation of treatment, LuT recipients, and longer exposures to suboptimal antiviral drug concentrations.38 In these cases, a genotypic resistance testing in order to detect specific mutations in the UL97 (encoding a viral kinase that catalyzes the first step in the triphosphorylation of GCV) and UL54 (encoding the viral DNA polymerase) genes is warranted.32,33 Nevertheless, it must be remarked that resistance mutations are not always detected,38 and that these patients will likely benefit from reduction of immunosuppression and optimization of antiviral treatment. Oral VGCV can be switched to IV GCV and dose be increased to 7.5 or 10.0mg/kg/12h in recipients with normal renal function and without neutropenia.32,33
There are limited options (only supported by low-level evidence) regarding the management of CMV antiviral resistance, which must be guided by the results of the genotypic testing.32,33 Foscarnet is the drug of choice for mutations in the UL97 gene that confer high-level GCV resistance (M460V/I, H520Q, C592G, A594V, L595S and C603W mutations).30 Close monitoring of renal function and electrolyte balance is needed during the course of foscarnet therapy.30 Mutations in the UL54 gene can confer cross-resistance to GCV, foscarnet and the nucleoside phosphonate cidofovir (even in the absence of previous exposure to these drugs).32 Tapering of immunosuppression or conversion to mTOR inhibitor-containing regimen may be considered by carefully balancing the risk of graft rejection.32,33 Furthermore, guidance by an infectious diseases specialist is mandatory.33 Other options would include the use of artemisinin derivatives49 or leflunomide,50 although available experience is very limited.
Investigational strategies for the prevention and treatment of CMVLetermovir is a new orally bioavailable antiviral drug, which inhibits the pUL56 subunit of the viral terminase complex.51 It has been recently approved for anti-CMV prophylaxis in CMV-seropositive allo-HSCT recipients.51 A phase 3, randomized, double-blind trial comparing the efficacy and safety of letermovir versus VGCV for the prevention of CMV disease among high-risk KT is currently underway and estimated to be completed in 2021 (ClinicalTrials.gov NCT03443869). Letermovir has been occasionally used off-label for the treatment of GCV-resistant CMV infection after SOT. The experience in four recipients diagnosed with CMV retinitis has been recently described.52 Although all of them showed clinical and fundoscopic improvement, three patients failed to achieve virologic suppression (with demonstration of letermovir-resistant mutations in the UL56 gene in two).52 The emergence of the UL56 mutation C325Y while on therapy was also reported in a LuT recipient treated with letermovir for GCV-resistant CMV viremia.53 In view of these preliminary experiences suggesting a low genetic barrier to resistance, letermovir will most likely be restricted for the treatment of low viral loads and as prophylaxis, particularly in patients that cannot receive VGCV due to hematologic toxicity. In addition, it must be kept in mind that letermovir lacks activity against non-CMV herpesviruses like VZV or HSV and that shows significant drug interactions with cyclosporine.
Maribavir is a benzimidazole L-riboside that prevents viral encapsidation and nuclear egress of viral particles though competitive inhibition of ATP binding to the UL97 protein kinase.54 Although an early placebo-controlled dose-ranging trial performed in allo-HSCT recipients after engraftment reported a reduction in the incidence of CMV infection,55 a larger phase 3 study failed to demonstrate significant differences in the occurrence of CMV disease between maribavir (at a dose of 100mg every 12h) and placebo. In a similar way, the non-inferiority of maribavir as compared to oral GCV for the prevention of CMV disease in D+/R− LT recipients was not established in a later trial. In fact, the incidence of CMV disease or infection was actually lower with oral GCV than with maribavir at 100 days and 6 months.56 These discouraging results raised concerns about the inadequacy of the maribavir dose used. A recent phase 2, open-label study compared various doses of maribavir (400, 800 and 1200mg every 12h) versus VGCV as preemptive therapy in allo-HSCT and SOT recipients.57 Notably, at a dose of at least 400mg twice daily, maribavir showed a similar efficacy to that of VGCV for clearing CMV viremia, with a lower rate of neutropenia. Dysgeusia was the most relevant adverse event in the maribavir arm.57
Recent years have witnessed major developments in the implementation of immune monitoring strategies based on the serial assessment of CMV-specific cell-mediated responses, ultimately aimed at guiding and individualizing prevention strategies.58,59 Most of these techniques hinge on the enumeration or functional profiling of interferon-γ (IFN-γ)-producing CD4+ and CD8+ T-cells upon antigenic stimulation. In addition to the intracellular cytokine staining by flow cytometry, which is usually considered the “gold standard”, there are various commercially available assays. The enzyme-linked immunosorbent assay (ELISA)-based QuantiFERON-CMV® test (Qiagen GmbH, Hilden, Germany) detects IFN-γ release (measured in IU/mL) in whole blood by means of ex vivo stimulation with a pool of viral peptides (pp50, pp65, pp28, immediate early [IE]-1, IE-2 and glycoprotein B [gB]).60 Enzyme-linked immunosorbent spot (ELISpot) assays (T-Track® CMV, Lophius Biosciences GmbH, Regensburg, Germany and T-SPOT.CMV®, Oxford Immunotec, Abingdon, UK) quantify the production of IFN-γ in terms of spot forming units in a given number of peripheral blood mononuclear cells.61,62 Finally, the development of the major histocompatibility complex (MHC)-tetramer technology (Dextramer CMV® Kit, Immudex ApS, Copenhagen, Denmark) offers the opportunity of combining surface and intracellular phenotyping with functional assays, although it requires the knowledge of individual HLA types.63 These techniques may be applied to various clinical scenarios, such as the baseline stratification of R+ patients,64–66 the expected occurrence of CMV disease following cessation of prophylaxis among high-risk patients,67 the individualization of duration of VGCV prophylaxis,68 or the probability of spontaneous clearance of asymptomatic viremia in intermediate-risk patients.69
Adoptive immunotherapy is also an experimental approach that could be eventually useful in SOT recipients with refractory or GCV-resistant CMV disease. A recent single-arm phase 1 trial included 13 recipients that received in vitro-expanded autologous CMV-specific T-cells.70 Eleven patients showed clinical improvement with complete resolution or reduction in CMV viremia. Moreover, four of them experienced a sustained improvement in the functional quality of CMV-specific T-cell responses upon completion of therapy, with an increased frequency of T-cells expressing IFN-γ and CD107. No grade 3–5 adverse events were noticed, nor a deleterious impact on graft outcomes.70 Nevertheless, further controlled trials are needed to confirm the efficacy and clinical feasibility of this therapy.
CMV vaccines could also be a valuable tool for minimizing the risk of CMV infection after SOT, although pivotal clinical data is still pending. HB-101 is a bivalent vaccine that contains two replication-defective recombinant lymphocytic choriomeningitis virus vectors expressing pp65 and a truncated isoform of the viral gB. A randomized placebo-controlled phase 2 trial is currently recruiting adult CMV-seronegative patients listed to receive a renal graft from a living CMV-seropositive donor (ClinicalTrials.gov NCT03629080). The study should be completed by 2021. A similarly designed trial aimed at evaluating the efficacy and safety of a different vaccine (ASP0113) has already recruited 150 patients and is expected to be finished in 2020 (ClinicalTrials.gov NCT01974206).
EBVEBV is a widely disseminated herpesvirus belonging to the Gammaherpesvirinae subfamily which spreads through the contact of respiratory secretions from asymptomatic virus shedders to susceptible subjects.71 The majority of primary EBV infections occur during the first years of life and are generally subclinical.71 The virus preferentially infects naïve B-cells in Waldeyer's ring, leading to initiation of the latent phase. Approximately 90–95% of adults have specific IgG antibodies against the EBV nuclear antigen (EBNA), thus indicating previous infection. Like most members of the herpesvirus family, EBV possess the ability to maintain lifelong latency following primary infection and reactivate in the presence of impaired T-cell immunity.72 In immunocompetent children and young adults EBV is the causative agent of infectious mononucleosis, which is usually benign and has a limited course. In the SOT setting, EBV-infected B-cells can become lymphoproliferative blasts upon activation of certain gene transcription programs, resulting in post-transplant lymphoproliferative disorder (PTLD), a devastating complication that accounts for up to one fifth of all post-transplant de novo malignancies and carries a mortality rate close to 50%.73 Although this complication is much more common among EBV-seronegative pediatric patients that experience primary infection, PTLD can also arise in adult SOT recipients.74
Prevention strategies for EBV-related PTLDThe risk of EBV-positive PTLD depends on several factors: (a) type of SOT performed (with SBT and KT recipients facing the highest and lowest risk, respectively, as consequence of differences in the net state of immunosuppression and the amount of lymphoid tissue contained in the graft); (b) EBV D+/R− serostatus (a combination almost universally resulting in the donor-derived infection of the recipient); and (c) time elapsed since transplantation, which reflects accumulated immunosuppression (i.e. induction therapy, maintenance regimen and treatment for graft rejection episodes).71,73 Post-transplant primary infection among EBV-seronegative patients is also possible when non-leukodepleted blood products are used. The use of the T-cell coestimulation inhibitor belatacept has been associated with an increased risk of PTLD with central nervous system (CNS) involvement.75
EBV viral load should be routinely monitored in whole blood or plasma samples in all EBV-seronegative recipients at transplantation.76 In addition, EBV D+/R− patients would benefit from monitoring at weekly or biweekly intervals through the first post-transplant year, whenever possible, until EBV DNAemia is detected. Thereafter, weekly monitoring should be maintained during the initial acute phase of infection and then less frequently once an EBV viral load “set point” is achieved. EBV D−/R− patients should undergo monthly surveillance for EBV DNAemia during the first year after trasplantation.76 Ongoing monitoring beyond that point can be considered in patients with fluctuating immunosuppression, those receiving anti-rejection therapy, or as long as the viral “set point” has not yet achieved. EBV R+ patients do not require routine EBV DNAemia monitoring, except for SBT recipients and those undergoing retransplantation or with a previous diagnosis of PTLD.76
Reduction of immunosuppression in the presence of significantly increasing EBV viral loads constitutes the cornerstone for the prevention of EBV-associated PTLD, with various progressive step-wise schedules proposed in the literature that usually advocate for maintaining calcineurin inhibitor levels at lower therapeutic ranges.76,77 However, no universal protocols can be currently proposed to guide clinicians through this process, and the evidence supporting the benefit of conversion to mTOR inhibitor-containing regimens is limited. Monitoring of graft function is essential during this period to allow early diagnosis of rejection. There is no firm evidence to suggest that GCV or VGCV therapy would have a role in EBV-seropositive recipients with increasing EBV viral load kinetics.77 However, some experts consider the use of these antivirals as an adjunct to the tapering of immunosuppression to reduce de novo B-cell infection and recruitment into lymphoproliferation.77 Likewise, the efficacy of preventive therapy based on rituximab (an anti-CD20 monoclonal antibody) for SOT recipients who do not respond to immunosuppression reduction remains to be assessed, although most experts advocate for its use in the presence of rapidly increasing viral loads.76 It should be noted that notable heterogeneity in clinical practices has been observed across transplant centers.78
Treatment of EBV-related PTLDSuccessful treatment of PTLD relies on the prompt diagnosis at early stages of disease. This complication must be suspected in asymptomatic recipients with a significant increase (e.g. more than tenfold or >1log10copies/mL from baseline levels) in EBV viral load or in the presence of persistent symptoms and signs suggestive of mononucleosis-like disease or lymphoproliferative syndrome.77 Noteworthy, PTLD can develop in patients with undetected EBV DNAemia.
Treatment of established PTLD usually comprises a combination of several approaches, including reduction of immunosuppression, surgical extirpation of localized disease or local radiation therapy, rituximab monotherapy and chemotherapy.73,76
As previously noted, there is no a common recommendation for the tapering of immunosuppression, although a 25–50% reduction in trough levels of calcineurin inhibitors (cyclosporine or tacrolimus) and discontinuation of antimetabolite agents (azathioprine or mycophenolic acid) should be implemented whenever possible.79 Nonglucocorticoid immunosuppressive agents should be promptly stopped in critically ill patients with extensive or life-threatening disease. Clinical response is expected to occur within the first 2–4 weeks, a delay that limits its sole use in aggressive forms of PTLD.79
Rituximab is considered a standard therapy for patients with nondestructive CD20-positive B-cell PTLD (plasmacytic hyperplasia, infectious mononucleosis-like PTLD, and florid follicular hyperplasia), polymorphic PTLD, or monomorphic diffuse large B-cell lymphoma (DLBCL)-like PTLD that have not responded to the reduction of immunosuppression.73 Rituximab can also be combined with chemotherapy in non-DLBCL, CD20-positive monomorphic PTLD subtypes. Nonetheless, it must be kept in mind that rituximab therapy can induce serious side effects, such as tumor lysis-like syndrome, prolonged HGG, bowel perforation, CMV reactivation or even progressive multifocal leukoencephalopathy.80
Chemotherapy is indicated for nondestructive PTLD, polymorphic PTLD, or monomorphic DLBCL-like PTLD if no response is achieved with the previous approaches.73,76 In these cases, the protocol used in the RSST trial (R-CHOP 21) appears to safe and effective for adult patients.81 Cytotoxic regimens are also recommended for other non-DLBCL monomorphic subtypes.73,76
Investigational strategies for the prevention and treatment EBV-associated PTLDInfusion of EBV-specific cytotoxic T-cells82 or radioimmunotherapy with 90Y-ibritumomab tiuxetan (a conjugate monoclonal antibodies targeting CD20)83 have preliminarily shown favorable results in small series of SOT recipients with PTLD, although further prospective studies are necessary. Future efforts should be directed to the standardization of EBV DNAemia monitoring practices, management of maintenance immunosuppression, and preemptive use of rituximab.
VZVPrimary VZV infection is uncommon following adult SOT since most recipients are VZV-seropositive at transplantation. Therefore, the majority of episodes take the form of herpes zoster (HZ) or shingles attributable to VZV reactivation, which is associated with older recipient age, induction therapy with lymphocyte-depleting agents, intensive immunosuppressive regimens, low natural killer cell counts and lack of anti-CMV prophylaxis with VGCV.11,84,85 HZ can appear at any time during the post-transplant period, although most cases occur beyond the first year. Incidence rates vary according to the SOT population, being higher among HT and LuT recipients.86,87 In addition, the development of post-herpetic neuralgia has been shown to be more common than in the general population.72
Prevention strategies for VZV infectionAll VZV-seronegative SOT candidates must receive the live attenuated Oka/Merck VZV strain-containing vaccine (Zostavax®, Merck Sharp & Dohme) at least 4 weeks prior to transplantation, as long as no contraindications are present (Table 3). It is also recommended to vaccinate family members, caregivers and others household members who are VZV-seronegative.88 If a varicella-rash develops following vaccination, the SOT recipient should be isolated and avoid contact with the household member due to the risk of transmission of the vaccine strain. Live attenuated vaccines are formally contraindicated after SOT since severe and often life-threatening complications have been reported with their use.88
Recommendations for the prevention of VZV reactivation in adult SOT recipients.
| Measure | Pre-transplant period | Post-transplant period |
|---|---|---|
| Live attenuated virus vaccine | If VZV-seronegative candidates, at least 4 weeks prior to transplantation | Contraindicated |
| Acyclovira,b | N/A | 600–1000mg daily in 3–5 doses |
| Valacyclovira,b | N/A | 500mg every 12h |
| Post-exposure prophylaxis | ||
| Non-specific IVIGc | If seronegative candidates | If seronegative recipients |
| Acyclovir | 800mg PO every 6h, 10mg/kg IV every 8h | 800mg PO every 6h, 10mg/kg IV every 8h |
| Valacyclovir | 1g every 8h | 1g every 8h |
IV: intravenous; IVIG: polyclonal intravenous immunoglobulin; N/A: not applicable; PO: oral route; VZV: varicella zoster virus.
Patients receiving ganciclovir or valganciclovir for CMV prophylaxis are protected against VZV reactivation.
As previously mentioned, CMV D−/R− recipients who are seropositive for HSV and/or VZV may benefit from a short prophylactic course with acyclovir or valacyclovir. The exposure of a susceptible (i.e. VZV-seronegative) recipient to a patient diagnosed with varicella carries a significant risk of developing primary infection since the virus can be transmitted by direct contact with vesicles and blisters or through airborne particles. Although controlled trials in this scenario are lacking, post-exposure prophylaxis based on a combination of non-specific IVIG and acyclovir or valacyclovir could be useful.88 IVIG therapy should be administered as soon as possible and always within the first 10 days from exposure. Two different antiviral prophylaxis regimens have been proposed: a shorter 7-day course (beginning at days 7–10 after exposure) and a longer regimen from days 3 to 22 (or from days 3 to 28 if IVIG is administered).
Treatment of VZV infectionPatients diagnosed with varicella (primary VZV infection) must be treated with IV acyclovir within the first 24h from the onset of the rash (Table 4). Parenteral therapy can be switched to oral therapy as soon as the patient has clinically improved and the lesions have crusted over. Since oral acyclovir has an unpredictable absorption and low bioavailability, valacyclovir or famciclovir are preferred. The use of non-specific IVIG is not routinely recommended.88
Recommendations for the treatment of VZV reactivation.
| Clinical scenario | Recommendation | Evidence |
|---|---|---|
| Localized HZ (single dermatomal) | Acyclovir 800mg PO five times daily,avalacyclovir 1g PO every 8h, famciclovir 500mg PO every 8ha | Strong, moderate |
| Varicella | Acyclovir 10mg/kg IV every 8hb,c | Strong, low |
| Disseminated or invasive HZHZ ophthalmicusdHZ oticus or Ramsay-Hunt syndromee | Acyclovir 10mg/kg IV every 8hb,c | Strong, moderate |
HZ: herpes zoster; IV: intravenous; PO: oral route; VZV: virus zoster varicella.
Treatment for HZ depends on the extension of the episode.88 In case of localized HZ with involvement of a single dermatome, orally administered acyclovir, valacyclovir or famciclovir are suitable treatment options. Most SOT recipients will not require hospital admission but should be closely followed. In the case of disseminated or organ invasive HZ, HZ ophthalmicus or HZ oticus (Ramsay-Hunt syndrome), IV acyclovir for at least 7 days is mandatory. Ophthalmology consultation is recommended whenever ophthalmic involvement is present. Foscarnet or cidofovir are alternative agents in patients who are allergic to acyclovir or have documented antiviral-resistance.88
Investigational strategies for the prevention and treatment of VZVShingrix® (GlaxoSmithKline) is an inactivated recombinant HZ subunit vaccine (HZ/su) containing the VZV glycoprotein E (gE) and the adjuvant system AS01B. It has been approved by the Food and Drugs Administration (FDA) in October 2017 for the prevention of HZ and post-herpetic neuralgia in VZV-seropositive patients 50 years or older. The European Medicines Agent (EMA) granted approval in March 2018 and is expected to be available in Spain by 2020. It is foreseeable that the HZ/su vaccine will be useful for preventing post-transplant VZV reactivation, but evidence is still scarce. Two clinical trials are currently underway to assess the safety and immunogenicity of this vaccine in the SOT population. A phase 4 trial will compare two vaccination schedules (receipt of the first HZ/su dose at 3–6 months versus 12–36 months after transplantation) among KT recipients (ClinicalTrials.gov NCT03993717), whereas a second study is recruiting LuT recipients to be vaccinated before and after transplantation (ClinicalTrials.gov NCT03493776).
HSV-1 and HSV-2The overwhelming majority of adult SOT recipients are seropositive for HSV-1 and/or HSV-2 at the moment of transplantation. Nonetheless, these patients face high rates of HSV reactivation as compared to the non-transplant population, with an increased risk of developing severe complications, and respond poorly to antiviral therapy.11
Prevention strategies for HSV reactivationAccording to the recently published guidelines from the AST Infectious Diseases Community of Practice,89 acyclovir, valacyclovir or famciclovir should be considered as antiviral drugs for HSV-1/2 prophylaxis among seropositive SOT recipients with no indication for anti-CMV prophylaxis (i.e. CMV D−/R− recipients). Recommended doses for HSV prevention and treatment in patients with normal renal function are detailed in Table 5. Prophylaxis should be maintained for at least the first month of transplantation and may be resumed in patients treated with lymphocyte-depleting agents for rejection episodes, since an increased risk of HSV recurrence has been observed in such scenario.89 Behavioral counseling remains the cornerstone for the prevention of HSV-1 and HSV-2 primary infection in seronegative patients. Contact with subjects with active herpetic lesions must be avoided, and HSV-2-seronegative recipients should consider having their partner tested.89 Daily valacyclovir therapy has been shown to reduce by 48% the rate of HSV-2 transmission in HSV-2-serodiscordant immunocompetent heterosexual couples, likely by decreasing the frequency and amount of HSV-2 shed in the genital tract by the HSV-2-infected partner.90 However, this strategy has never been evaluated in the specific SOT setting.
Recommendations for the prevention and treatment of HSV-1 and HSV-2 reactivation in adult SOT recipients.
| Clinical scenario | Recommended drugs |
|---|---|
| Prophylaxisa,b | Acyclovir 400–800mg PO every 12h,valacyclovir 500mg PO every 12h,famciclovir 500mg PO every 12h |
| Localized mucocutaneous diseasec | Acyclovir 400mg PO every 8h,acyclovir 5mg/kg IV every 8h,dvalacyclovir 1g PO every 12h,famciclovir 500mg PO every 12h |
| Disseminated mucocutaneous diseaseVisceral or CNS involvement | Acyclovir 10mg/kg IV every 8he |
| Acyclovir-resistant HSVf | Foscarnet 80–120mg/kg IV in 2–3 divided doses, cidofovir 5mg/kg IV weekly given with probenecid |
CNS: central nervous system; HSV: herpes simplex virus; IV: intravenous; PO: oral route.
Patients receiving ganciclovir or valganciclovir for CMV prophylaxis are protected against HSV reactivation.
Recommended for at least the first month after transplantation and following treatment of rejection episodes.
Due to the potential severity of HSV infection in SOT recipients, treatment should be started as soon as clinical suspicion is raised and not be delayed until microbiological confirmation.89 Limited mucocutaneous disease can be safely treated with oral acyclovir, valacyclovir or famciclovir, which should be continued until complete remission of the herpetic lesions and for a minimum of 5–7 days, whereas IV acyclovir (at 5mg/kg every 8h) would be reserved to those in whom oral administration is not feasible (Table 5).89 Cases of disseminated mucocutaneous disease, visceral infection (e.g. hepatitis) or CNS involvement require IV acyclovir at 10mg/kg every 8h for 14 days (except in the presence of CNS involvement, in which a 21-day course is recommended). Reduction of immunosuppression should be considered in cases of severe HSV disease, whenever possible.89
In contrast to immunocompetent patients, mutations in the thymidine kinase gene may confer acyclovir resistance among allo-HSCT and, at a lower incidence, SOT recipients, rendering conventional dosing regimens of acyclovir and its analogs ineffective. Risk factors in the allo-HSCT population include prolonged acyclovir exposure and the development of graft-versus-host disease.91 In these cases, acyclovir must be empirically switched to foscarnet or cidofovir and a genotypic testing should be requested.89 There have been also successful experiences with the use of high-dose IV acyclovir by continuous infusion.92
Long-term suppressive acyclovir therapy may be considered for recipients with two or more HSV recurrences following discontinuation of prophylaxis and maintained, ideally, until immunosuppression can be reduced. Suppressive therapy is associated with a lower risk of inducing acyclovir resistance than repeated course of treatment for recurrent episodes. Nonetheless, acyclovir toxicity and cost, recipient's adherence and psychosocial issues should be taken into account by the treating clinician.89
BKPyVBKPyV is one of the 14 human polyomaviruses within the Polyomaviridae family, which is composed of more than 70 species.93 BKPyV infection usually occurs within the first decade of life, with seroprevalence rates progressively increasing up to 90% in the adult general population.94 Infection is thought to occur via the respiratory or the oral route.94 Following primary viremia, BKPyV reaches the renourinary tract where the virus is capable of maintaining latent infection. BKPyV may be detected in urine samples of healthy subjects and various SOT populations, although the risk of progression from BKPyV viruria to viremia and BKPyV-induced disease is almost exclusively restricted to KT recipients.26 In fact, BKPyV-associated nephropathy (BKPyVAN) currently constitutes one of the leading causes of renal graft loss,94 whereas the attributable effect on renal function among non-KT recipients is questionable.26
Prevention strategies for BKPyV infectionSeveral risk factors have been identified for the development of BKPyVAN after KT, such as deceased donor, older recipient age and male gender, number of human leukocyte antigen mismatches, cold ischemia time, use of ureteric stents and tacrolimus-bases immunosuppression regimens (as compared to cyclosporine). Since effective and safe antiviral agents with activity against BKPyV are still lacking, the cornerstone for the prevention of BKPyVAN is screening for BKPyV viremia with preemptive reduction of immunosuppression if necessary.95 It is recommended that BKPyV DNAemia detection should to be performed monthly until post-transplant month 9 and every 3 months thereafter until the second year.95 BKPyV DNAemia should be also requested at the time of graft biopsy in cause of graft dysfunction. This strategy is thought to identify up to 90% of KT recipients at risk of BKPyVAN before significant graft function impairment occurs. Since fluoroquinolones can inhibit BKPyV DNA topoisomerase, some retrospective single-center studies reported that the use of levofloxacin or ciprofloxacin as prophylaxis for urinary tract infection (even as a short one-month course) may reduce the occurrence of BKPyV viremia.96,97 Nevertheless, a placebo-controlled, double-blinded, randomized trial failed to demonstrate a significant impact of a 3-month course of levofloxacin in terms of BKPyV viruria or viremia.98
Treatment of BKPyV infectionThe primary approach to significant BKPyV DNAemia (>10,000copies/mL) and established BKPyVAN consists of reduction of immunosuppression (of note, acute or chronic graft rejection must have been previously ruled out). Tacrolimus trough levels are usually targeted to<6ng/mL, cyclosporine levels to<150ng/mL, sirolimus levels to<6ng/mL, and mycophenolic acid daily dose equivalents reduced to half or less of the daily maintenance dose. Further measures include switching from tacrolimus to reduced-exposure cyclosporine or conversion to mTOR inhibitor-containing regimens.95 A 30-day course of levofloxacin associated to reduction of immunosuppression was not proven to significantly reduce BKPyV viral load in a placebo-controlled trial.99 Cidofovir, leflunomide or replacement with IVIG preparations containing high titers of anti-BKPyV neutralizing antibodies have been also used,95 although no trials support its efficacy. Brincidofovir (CMX-001) is a novel, broad-spectrum lipid antiviral conjugate comprised of a lipid (1–0-hexadecyl-oxypropyl) covalently linked to the phosphonate group of cidofovir, which has been proven to exert a potent in vitro activity against BKPyV in primary human urothelial cells.100,101 Unfortunately, the clinical development program of this agent has been temporarily stopped, and results from a placebo-controlled trial assessing the use of brincidofovir for BKPyV viruria in KT and HSCT recipients (ClinicalTrials.gov NCT00793598) remain unpublished. Moreover, it has been reported the development of brincidofovir-induced reversible severe acute kidney injury in two SOT recipients infected with UL97-mutant CMV strains.102
Investigational strategies for the prevention and treatment of BKPyVStandardization of BKPyV PCR assays providing results in IU/mL are needed in order to harmonize clinical procedures across centers and reporting of study results.95 Strategies for immunosuppression tapering or switching should be ideally evaluated in the setting of clinical trials. Finally, data is also lacking concerning the potential value of adoptive therapy with BKPyV-specific T-cells.
ConclusionsThe implementation over the past decades of risk minimization strategies has contribute to substantially reduce the negative impact of post-transplant viral infections (namely CMV, VZV and HSV-1/2) on graft and patient outcomes, although this complication persists as a major concern for transplant infectious diseases physicians. Future developments in antivirals drugs, vaccine research, strategies for immune monitoring and adoptive immunotherapy will most surely open new prospects that will eventually improve long-term results of the SOT procedure.
Authors’ contributionsJ.T.S. was responsible for the literature review and the drafting and writing of the manuscript; M.F.R. and J.M.A. contributed to the critical revision of the manuscript and approved the final version.
Funding sourcesM.F.R. holds a “Miguel Servet” research contract (CP18/00073) from the Instituto de Salud Carlos III, Spanish Ministry of Science, Innovation and Universities.
Conflict of interest statementThe authors have no conflicts of interest related to this publication.
