




Las enfermedades infecciosas tienen gran incidencia en la población, provocando un impacto en la salud mundial. La primera técnica aplicada al diagnóstico de las infecciones es el cultivo de los microorganismos in vitro, el cual es costoso y con un resultado tardío. En las últimas décadas los esfuerzos se han centrado en la aplicabilidad de las ciencias «ómicas», destacando el avance proporcionado por las técnicas proteómicas en el campo de las enfermedades infecciosas. La presente revisión expone el manejo, el procesamiento y el análisis de las muestras biológicas para su estudio proteómico.
Infectious diseases have a high incidence in the population, causing a major impact on global health. In vitro culture of microorganisms is the first technique applied for infection diagnosis which is laborious and time consuming. In recent decades, efforts have been focused on the applicability of «Omics» sciences, highlighting the progress provided by proteomic techniques in the field of infectious diseases. This review describes the management, processing and analysis of biological samples for proteomic research.
Las enfermedades infecciosas comportan uno de los mayores problemas de salud a nivel mundial, provocando una gran morbimortalidad y alcanzando niveles de millones de afectados cada año1. El incremento de viajes, cambios demográficos y resistencias a antimicrobianos contribuye significativamente a este fenómeno. El tratamiento de las enfermedades infecciosas se basa en un primer diagnóstico seguido de la administración del fármaco correspondiente.
La mayoría de las enfermedades infecciosas son la consecuencia de una compleja red de interacciones que pueden implicar de cientos a miles de factores, tanto del patógeno como del hospedador. Para alcanzar un mejor entendimiento de los procesos de las enfermedades infecciosas es esencial realizar estudios traslacionales y multidisciplinares donde se integran la cooperación del personal implicado en las diferentes áreas de trabajo.
Los análisis más exhaustivos realizados hasta el momento en cuanto a la búsqueda de biomarcadores y nuevas dianas para fármacos y vacunas se han realizado a partir de estudios en genómica y transcriptómica. En las últimas décadas ha aumentado significativamente el número de estudios proteómicos en enfermedades infecciosas y en todo el ámbito clínico para la búsqueda de biomarcadores y nuevas dianas para la vacunación y fármacos.
La secuenciación del genoma microbiano ha revolucionado el estudio de los patógenos, no solo a nivel de microbiología básica, sino también en diagnóstico, epidemiología, fisiopatología, tratamiento de las enfermedades y desarrollo de vacunas. Además, la secuenciación del genoma microbiano ha posibilitado el estudio de sus proteomas. El primer genoma secuenciado fue el virus del Epstein-Barr (1984), de 170Kbs2, y una década después, en 1995, se secuenció el genoma de la bacteria gramnegativa Haemophilus influenzae, de 1,8Mb3.
Muchos estudios se basaban en el análisis de los genes expresados en diferentes tipos celulares y tejidos en diferentes contextos fisiológicos, principalmente mediante un análisis de ARN mensajero (ARNm), sin existir a menudo una correlación directa entre el contenido en ARNm y el contenido proteico. La falta de correlación entre los niveles ARNm-proteína se debe a que el conjunto de proteína producida por una cantidad dada de ARNm depende del estado fisiológico de la célula4. Un ejemplo de esta ausencia de correlación entre transcriptómica (ARNm) y proteómica (proteína) se demuestra en el trabajo de Franco et al.5, donde se comparan dos cepas de Helicobacter pylori, una carcinogénica y un progenitor no carcinogénico. Los análisis en ARNm no mostraron diferencias entre ambas cepas, mientras que a nivel proteico estas cepas eran diferenciables.
La proteómica es considerada el siguiente paso en el estudio de un sistema biológico, después de la genómica y transcriptómica. La complejidad de los estudios proteómicos es mayor, ya que el genoma de un organismo es estático, mientras que el proteoma difiere de una célula a otra y entre estados. El término proteoma6, definido por Marc Wilkins en 1994, es una imagen dinámica de todas las proteínas expresadas por un organismo, célula o compartimento subcelular concreto, en un momento dado y bajo determinadas condiciones, constituyendo el mapa de expresión proteica de una célula, tejido u organismo dado. Un factor adicional de complejidad son las modificaciones que puede experimentar la estructura o secuencia básica de la proteína (p.ej., el procesado proteolítico) y las modificaciones postraduccionales (PTM), que sirven para modificar o modular la actividad, función o localización de una proteína en diferentes contextos fisiológicos o metabólicos. Finalmente, otro factor de complejidad es el moonlighting o multifuncionalidad, en que una misma proteína puede realizar varias funciones7.
La proteómica, término también acuñado por Wilkins, es un nuevo enfoque en el estudio de las proteínas que ha evolucionado en los últimos años gracias a la integración de tecnologías como métodos de separación de proteínas de alto rendimiento, espectrometría de masas (MS) y, sobre todo, herramientas en bioinformática que han permitido el análisis de un gran volumen de datos. Gracias a su aplicabilidad, la proteómica permite no solo identificar las proteínas, sino también categorizarlas y clasificarlas en cuanto a su función e interacciones.
El campo de estudio de la proteómica es realmente amplio, pero podemos agruparlo en 3 subgrupos: a)proteómica de expresión, cuyo objetivo es identificar las proteínas presentes en la muestra así como obtener toda la información acerca de abundancias, PTM y localización subcelular; b)proteómica estructural, que permite la obtención de la estructura tridimensional de las proteínas, y c)proteómica funcional, cuya finalidad es averiguar la función de las proteínas y conocer las interacciones que puedan efectuar.
Las técnicas en proteómica ofrecen un novedoso y potente enfoque para el diagnóstico de patógenos, seguimiento de brotes, dinámica patógeno-hospedador, control de la enfermedad y desarrollo de fármacos y vacunas.
La presente revisión tiene como objetivo mostrar la amplia variedad de técnicas y aplicaciones disponibles en el campo de la proteómica destinadas al estudio de las enfermedades infecciosas.
Metodologías proteómicasLas técnicas en proteómica envuelven multitud de metodologías experimentales, como electroforesis en 2 dimensiones (2DE), cromatografía líquida (LC) y MS. La combinación de estas técnicas con un posterior análisis bioinformático es utilizada para el estudio proteico de una gran variedad de organismos. Estas técnicas son potencialmente valiosas, aunque hay complejidades en las muestras que pueden afectar a su análisis e interpretación, como son el grado de PTM, el alcance dinámico de la expresión (p.ej., la abundancia de albúmina en plasma enmascara proteínas minoritarias) y problemas de detección de proteínas asociadas a membrana8.
El mayor obstáculo para el progreso en el área de la proteómica es la dificultad en la recopilación de patógenos in vivo en suficiente abundancia y de forma adecuada para análisis proteómicos. Aun siendo estas las condiciones idóneas para estudios de expresión de proteínas en microorganismos patógenos, existen casos en los que no se puede realizar y el cultivo axénico (in vitro) se convierte en la mejor opción. Se han analizado multitud de patógenos utilizando técnicas proteómicas en condiciones in vitro. Estos estudios permiten conocer la magnitud de expresión de proteínas y tener una idea global de su proteoma. Basarse únicamente en el estudio del proteoma en estas condiciones puede provocar la pérdida de una información fundamental9-12, ya que no permite identificar las proteínas implicadas en infección, como factores de virulencia, proteínas que permiten la evasión de la respuesta inmune del hospedador y proteínas que permiten la utilización de la maquinaria celular del hospedador para propagarse. Una simulación experimental de las condiciones de infección in vivo es la infección de cultivos celulares (simulación in vitro del entorno de infección) o la utilización de modelos animales13,14. La desventaja en estos estudios radica en la presencia de una gran «contaminación» por proteínas del hospedador, además de una baja obtención de biomasa del patógeno.
En conjunto, enfoques experimentales in vitro/ex vivo tienen implicaciones significantes con respecto a la identificación de nuevas vacunas, dianas para fármacos, biomarcadores de infectividad y progreso de enfermedad.
La mayoría de las proteínas implicadas en los primeros estadios de infección del hospedador son las proteínas de superficie celular. Estas proteínas son la conexión entre la célula y el medio extracelular, por lo que son el mediador principal en la infección. Por consiguiente, son componentes de gran interés para el desarrollo de nuevas vacunas y para la caracterización de nuevas dianas para fármacos. La información de estas moléculas se encuentra en baja representación en los estudios proteómicos debido a su baja abundancia, poca solubilidad y la problemática en su fraccionamiento sin una contaminación por proteínas citoplasmáticas15,16. En el caso del estudio de las bacterias grampositivas y hongos, también es importante conocer la composición proteica de la pared celular y no solo de la membrana plasmática17.
Preparación de muestraLa obtención y preparación de las muestras es uno de los pasos más relevantes en los estudios proteómicos. Es de vital importancia que las muestras sean tomadas y procesadas mediante el mismo protocolo para que los resultados obtenidos sean veraces, reproducibles y que las diferencias observadas no sean fruto de la manipulación.
El procesamiento de la muestra en el laboratorio difiere según la procedencia de esta y la finalidad del estudio. En los estudios en cultivo axénico no es necesario un aislamiento previo para la obtención de la muestra proteica. Por el contrario, cuando se analiza el microorganismo en estado infectivo, la preparación de la muestra precisa de un aislamiento del microorganismo respecto a las células del hospedador. Los métodos más comúnmente utilizados para la separación son la centrifugación, la separación inmunomagnética (IMS) y la clasificación de células mediante citometría de flujo (FACS).
El primer método, la centrifugación, es el recomendado para patógenos con pared celular (plantas, hongos, algas, arqueas y bacterias grampositivas), previa lisis de las células eucariotas (hospedador) por presión osmótica18,19 o detergentes como el tritónX-11413.
En el segundo método, la técnica de IMS, se utilizan partículas magnéticas conjugadas con una inmunoglobulina (Ig) específica frente al microorganismo de interés. Estos microorganismos pueden ser fácilmente enriquecidos y purificados, generando suficiente biomasa virtualmente libre de proteínas del hospedador para un exhaustivo análisis del proteoma. La ventaja más importante respecto a la centrifugación es la obtención de una muestra libre de contaminantes procedente del hospedador. Por el contrario, esta técnica requiere anticuerpos específicos frente a estructuras de la superficie del patógeno.
En el tercer método, el FACS, se utilizan anticuerpos específicos para el marcaje fluorescente del microorganismo diana, con la finalidad de facilitar la separación de este respecto a otros microorganismos o detritos de la célula hospedadora. Esta técnica puede separar miles de partículas por segundo, pero la acumulación de suficiente material patógeno para la aplicación de técnicas proteómicas requiere bastantes horas de clasificación, haciéndola muy costosa.
Todos estos métodos de separación deben ser optimizados para incrementar la velocidad de procesamiento de la muestra y reducir el riesgo de digestión parcial por proteasas, además de reducir la probabilidad de contaminación con proteínas del hospedador.
Los estudios en proteómica se pueden dirigir al estudio del proteoma total, subproteoma o una proteína concreta. La selección de una proteína o población proteica determinada se debe realizar mediante un fraccionamiento de la muestra. Entre las técnicas utilizadas para esta finalidad cabe destacar la purificación de afinidad en tándem (TAP)20, co-inmunoprecipitación (CO-IP)21 y el chip de exploración del peptidoma mediante HLA22,23.
Técnicas proteómicasLas técnicas proteómicas se dividen en cualitativas y cuantitativas. Las primeras informan acerca de la expresión o no de una determinada proteína o conjunto proteico (presencia/ausencia), mientras que la proteómica cuantitativa permite determinar y comparar la cantidad de proteína presente en la muestra.
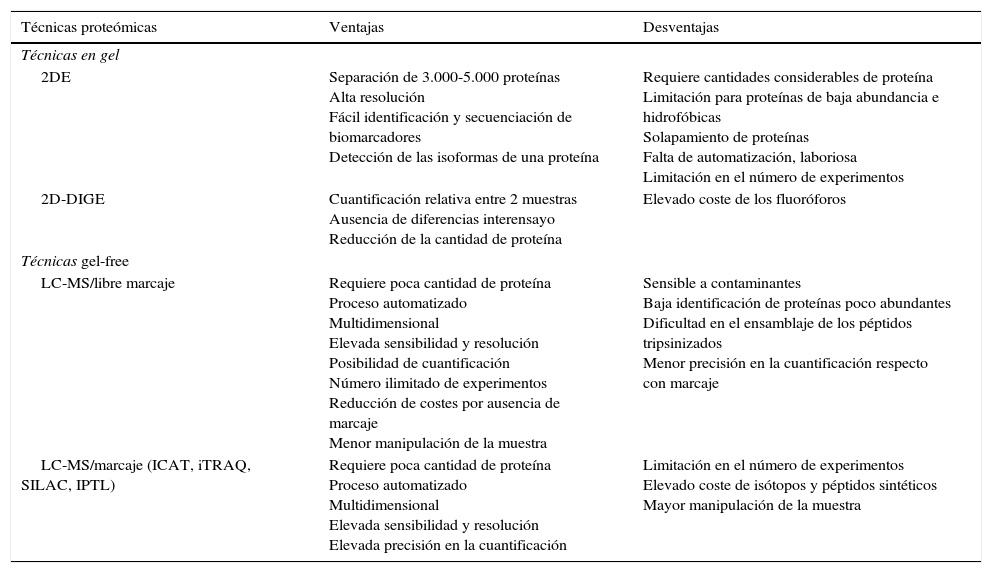
En los estudios en proteómica, la muestra es una mezcla compleja de cientos/miles de proteínas. Por este motivo es esencial la utilización de una técnica de separación. Estas técnicas se agrupan en: en gel y gel-free. Las técnicas en gel son aquellas en las que se utiliza un entramado de polímeros para la separación. Las técnicas gel-free realizan la separación de la muestra proteica en solución mediante diferentes tipos de cromatografía líquida (intercambio iónico, afinidad, fase reversa…). En la tabla 1 se muestran las ventajas y desventajas de las distintas técnicas proteómicas.
Resumen de las principales ventajas y desventajas de las técnicas en gel y gel-free de proteómica
| Técnicas proteómicas | Ventajas | Desventajas |
|---|---|---|
| Técnicas en gel | ||
| 2DE | Separación de 3.000-5.000 proteínas Alta resolución Fácil identificación y secuenciación de biomarcadores Detección de las isoformas de una proteína | Requiere cantidades considerables de proteína Limitación para proteínas de baja abundancia e hidrofóbicas Solapamiento de proteínas Falta de automatización, laboriosa Limitación en el número de experimentos |
| 2D-DIGE | Cuantificación relativa entre 2 muestras Ausencia de diferencias interensayo Reducción de la cantidad de proteína | Elevado coste de los fluoróforos |
| Técnicas gel-free | ||
| LC-MS/libre marcaje | Requiere poca cantidad de proteína Proceso automatizado Multidimensional Elevada sensibilidad y resolución Posibilidad de cuantificación Número ilimitado de experimentos Reducción de costes por ausencia de marcaje Menor manipulación de la muestra | Sensible a contaminantes Baja identificación de proteínas poco abundantes Dificultad en el ensamblaje de los péptidos tripsinizados Menor precisión en la cuantificación respecto con marcaje |
| LC-MS/marcaje (ICAT, iTRAQ, SILAC, IPTL) | Requiere poca cantidad de proteína Proceso automatizado Multidimensional Elevada sensibilidad y resolución Elevada precisión en la cuantificación | Limitación en el número de experimentos Elevado coste de isótopos y péptidos sintéticos Mayor manipulación de la muestra |
La separación de las proteínas en gel es una herramienta que permite la separación de estas por propiedades como el tamaño (Mr), el punto isoeléctrico (pI) y/o la conformación (geles nativos). La separación en gel se puede realizar por una sola propiedad (monodimensional) o por 2 propiedades consecutivas (bidimensional). Debido a la complejidad de los extractos proteicos totales, la separación de las muestras en electroforesis bidimensional (2DE) ofrece una mayor resolución. Mediante esta técnica las proteínas son separadas por pI y posteriormente por Mr, obteniendo un mapa global del proteoma de la muestra.
La técnica 2DE únicamente puede ser aplicada satisfactoriamente si hay un mínimo de 108 células aisladas. Esta técnica permite separar entre 3.000-5.000 proteínas únicas24, diferenciadas generalmente en un único spot. No obstante, una misma proteína puede identificarse en diferentes spots, indicando que esta proteína presenta varias isoformas que difieren en pI y/o Mr. Estas isoformas pueden informar acerca de las posibles PTM de una determinada proteína. Las PTM más frecuentes son fosforilaciones, glucosilaciones o proteólisis limitada25. Otro fenómeno menos común es la identificación de dos proteínas en el mismo spot, debido a su igual pI y Mr.
La preparación de la muestra para la electroforesis bidimensional es crucial para la obtención de resultados óptimos, siendo necesarios procesos de solubilización, disgregación, desnaturalización y reducción de las proteínas26.
Una posibilidad que ofrecen los geles 2D es su combinación con técnicas de marcaje de proteínas permitiendo la comparación de proteomas en un mismo gel, evitando de esta manera las variaciones interensayo (entre geles/experimentos). Esta técnica es conocida como gel bidimensional diferencial (2D-DIGE). El marcaje mediante fluoróforos puede ser aplicado al estudio comparativo de 2 proteomas permitiendo una cuantificación relativa de las muestras. Este marcaje puede dirigirse a conjuntos específicos de proteínas; por ejemplo, el estudio de proteínas con regiones expuestas hacia el medio extracelular. La aplicabilidad de esta técnica ha permitido el estudio del surfoma27 evitando el fraccionamiento de proteínas de superficie, las cuales suelen ser largas y costosas.
Los geles 2D-DIGE se pueden aplicar a la búsqueda de dianas terapéuticas analizando el proteoma in vitro frente al proteoma del patógeno en situación de infección, obteniendo en el mismo gel la comparativa del proteoma en ambas situaciones. Debido a la dificultad para obtener muestra suficiente de patógeno en situación de infección, se han realizado estudios en los que se compara el proteoma del microorganismo in vitro en diferentes fases de crecimiento. En el trabajo de Hayashi et al.28 se estudiaron las proteínas expresadas por Legionella pneumophila en 2 fases de crecimiento: exponencial y postexponencial, observándose una elevada expresión de factores de virulencia en la fase postexponencial. Gracias a este estudio se puede establecer en futuros trabajos en Legionella el proteoma en fase postexponencial como aproximación del proteoma en infección.
Una desventaja importante de la técnica 2DE es la limitación en la capacidad de enfoque de proteínas de media a baja abundancia debido a la gran gama de niveles de expresión de proteínas. Las proteínas más afectadas por dicha limitación son las proteínas de membrana y proteínas de bajo peso molecular, que se encuentran subrepresentadas en proteomas totales. Estas proteínas pueden ser mejor detectadas mediante otras técnicas proteómicas29.
Técnicas gel-freeLa técnica gel-free requiere una primera separación de las proteínas presentes en la muestra mediante cromatografía líquida por diferentes propiedades, definidas según la columna utilizada. Existen en el mercado diferentes tipos de columnas, siendo las más utilizadas las de fase reversa, intercambio iónico, afinidad y exclusión molecular. Posteriormente se realiza un análisis mediante MS para la identificación de las proteínas (LC-MS/MS).
Las técnicas de separación gel-free son aconsejables cuando la cantidad de muestra es escasa, como es el caso de experimentos en infección en los que se obtiene una baja cantidad de microorganismos. En estos casos, las técnicas gel-free MS son adecuadas por ofrecer una mejor sensibilidad, ya que permite monitorizar 500-600 proteínas desde únicamente 106 células14 (una cantidad 100 veces menor que en los geles 2D). Esta técnica permite la detección de proteínas difícilmente separables en 2DE, como proteínas muy hidrofóbicas, proteínas de membrana y proteínas con pI o Mr extremos, como son las proteínas ribosomales con un pI básico30-32. Un claro ejemplo en el que se demuestra esta mayor sensibilidad es el trabajo realizado en Mycoplasma genitalium, modelo de genoma y proteoma mínimo, en el que se estudió la fracción rica en proteínas de membrana mediante fraccionamiento por tritón114. El análisis de esta fracción se realizó tanto por 2DE como por LC-MS, obteniéndose una clara diferencia en cuanto a número de proteínas identificadas. Mediante 2DE se identificaron 49 proteínas, mientras que por LC-MS se identificaron 242 proteínas33.
Proteómica cuantitativaLa proteómica cuantitativa es una herramienta útil para los análisis proteómicos mediante LC-MS/MS34. Los métodos de cuantificación se pueden clasificar en cuantificación relativa/absoluta o con/sin marcaje.
Los métodos de cuantificación relativa (ICAT, iTRAQ, IPTL o SILAC) son utilizados para la comparación de abundancias de proteínas o péptidos entre muestras. Por otro lado, mediante el uso de péptidos sintéticos marcados isotópicamente se obtiene una cuantificación absoluta de los péptidos diana.
Proteómica cuantitativa mediante marcajeLas técnicas cuantitativas mediante marcaje utilizan compuestos isotópicos para un posterior análisis mediante MS, obteniendo una cuantificación relativa. El marcaje de las proteínas puede efectuarse durante el crecimiento celular o una vez realizada la extracción de estas. Las muestras a comparar son diferenciadas mediante marcaje isotópico. Según la técnica de marcaje diferenciamos: ICAT (isotope code affinity tagging: marcaje por afinidad con codificación isotópica)35, iTRAQ (multiplexed isobaric tagging chemistry: etiquetado isobárico multiplexado químico)36 e IPTL (isobaric peptide termini labeling: unión terminal isobárica del péptido)37.
La cuantificación relativa mediante marcaje permite la comparativa de 2 poblaciones según su crecimiento celular mediante SILAC (stable isotope labeling by/with amino acids in cell culture: marcaje isotópico estable de aminoácidos en cultivo celular). En esta técnica, los marcadores isotópicos no radiactivos son incorporados durante el crecimiento celular a las proteínas sintetizadas de novo38-41.
Las técnicas de cuantificación mediante marcaje tienen ciertas limitaciones, como es el incremento de la complejidad en la preparación de la muestra (por el tratamiento independiente de la muestra), la necesidad de una gran concentración de esta, los costes elevados de los reactivos, marcajes incompletos que distorsionan el resultado y la necesidad de software específico para la cuantificación.
Proteómica cuantitativa libre de marcajeLas estrategias libres de marcaje pueden ser utilizadas para cuantificación tanto relativa como absoluta. Estas técnicas alcanzan una mayor rapidez, resultados más limpios y un análisis simple de los resultados. Por este motivo son prometedoras en la cuantificación de proteínas poco abundantes debido a su alta sensibilidad, aunque para ser aplicadas se deben tener en cuenta ciertos criterios: a)es necesario disponer de espectrómetros de masas con gran precisión e instrumentos HPLC con un tiempo seguro de retención; b)debe ser aplicado suficiente material para determinar la concentración proteica antes de la medición por MS con la finalidad de aplicar cantidades equivalentes de péptidos, y c)el número de péptidos de la célula hospedadora debe ser reducido al mínimo para evitar falsos positivos.
La cuantificación de proteínas por este método se basa generalmente en 2 categorías: a)medición de los cambios de intensidad de los iones, ya sea por área de los picos o por la altura de estos en espectrometría, y b)recuento de espectros de las proteínas identificadas después del análisis MS/MS42-53.
Una mejora de la proteómica cuantitativa sin marcaje es la incorporación de péptidos sintéticos marcados isotópicamente como estándares internos. Estos péptidos permiten una cuantificación absoluta (AQUA: absolute quantification) de los péptidos de interés54.
Instrumentos para análisis e identificación proteica mediante espectrometría de masasLa identificación de proteínas se realiza en su gran mayoría mediante espectrómetros de masas. Estos instrumentos permiten la obtención de iones a partir de moléculas orgánicas, separándolos y detectándolos en función de su masa/carga (m/z). Los espectrómetros de masas constan de 3 componentes: fuente de ionización, analizador de masa y detector. Existen diferentes tipos de espectrómetros de masas según la combinación de los diferentes elementos. Los más comúnmente utilizados son:
- •
MALDI-TOF (Matrix-Assisted Laser Desorption/Ionization-Time Of Flight: desorción/ionización mediante láser asistido por matriz, «tiempo de vuelo»): utilizado comúnmente para la identificación de proteínas mediante huella peptídica y el estudio de los perfiles proteicos de una muestra e interacciones de proteínas. Cabe destacar el desarrollo de la técnica del «Imaging», que permite una visualización en 2 dimensiones de la distribución de metales traza, metabolitos, lípidos de superficie, péptidos y proteínas, directamente en los tejidos sin realizar una fijación o extracción previa55,56.
- •
SELDI-TOF (Surface Enhanced Laser Desorption Ionization-Time Of Flight: desorción/ionización mediante láser de superficie mejorada, «tiempo de vuelo»): este instrumento utiliza la misma tecnología que el MALDI-TOF incorporando la funcionalización de la placa (intercambio iónico, fase reversa, anticuerpos, ADN, otras proteínas, etc.) en la que se carga la muestra. La comparación de las proteínas retenidas en la placa entre diferentes muestras puede ser utilizada para la búsqueda de biomarcadores57.
- •
ESI (ElectroSpray Ionization: ionización por electrospray): este dispositivo es especialmente útil para la secuenciación peptídica, ya que permite la ionización de las macromoléculas fragmentándolas por los enlaces peptídicos (uniones más débiles), obteniendo de esta forma la secuencia aminoacídica56,58.
Los análisis proteómicos generan una gran cantidad de datos a analizar. El desarrollo de herramientas bioinformáticas, como la introducción de nuevos algoritmos, ha sido clave para manipular dichos conjuntos de datos. Este campo permite y facilita la manipulación de datos a gran escala, desarrollando programas y herramientas de búsqueda oportunas. Estas herramientas se han aplicado con gran éxito en el procesamiento de datos MS tanto en análisis de huella peptídica (identificación de proteínas), huella de fragmentación peptídica (identificación de la secuencia de péptidos) y secuenciación de novo.
Existen multitud de programas que permiten la identificación in sillico de proteínas. En estudios proteómicos en enfermedades infecciosas existen programas que permiten la identificación y caracterización de epítopos del patógeno que interaccionan con el sistema inmune del hospedador, inmunoma. Las interacciones patógeno-hospedador no se conocen en profundidad en muchas de las enfermedades infecciosas, por lo que estudios bioinformáticos pueden dirigir el estudio de estas interacciones.
AplicacionesProteoma de microorganismosLos estudios del proteoma total de los microorganismos patógenos son muy valiosos en cuanto a la información que se obtiene de la expresión proteica en una condición determinada. En otros estudios es más interesante obtener información de determinados subproteomas, como las proteínas expuestas en la membrana, o conocer qué proteínas desencadenan la respuesta inmune en el hospedador. Cabe destacar el papel de las PTM en la interacción entre el patógeno y el hospedador, por lo que debería tenerse en cuenta su estudio en microorganismos patógenos.
Proteínas de membrana, de superficie y secretadasEl subproteoma de membrana constituye un conjunto de proteínas relacionadas con la infección. Existen técnicas para la detección de las proteínas expuestas en la superficie celular (surfoma), como son el marcaje específico mediante fluoróforos en el 2D-DIGE y el afeitado celular. La técnica del afeitado celular se ha utilizado satisfactoriamente en diversos patógenos humanos grampositivos como en Streptococcus59 o en Staphylococcus aureus60, aunque aún no se conoce con certeza su aplicabilidad en bacterias gramnegativas. Estas bacterias poseen una envuelta celular menos rígida y menos resistente que las grampositivas, por lo que el «afeitado» compromete la integridad de la membrana y provoca la lisis celular. Como alternativa a estas técnicas se han mejorado los protocolos de extracción de proteínas de membrana existentes a través de diferentes detergentes o aislamiento de membranas61, como es el caso de la utilización del tampón carbonato para la eliminación de proteínas solubles contaminantes en las muestras.
Las proteínas secretadas constituyen un importante subproteoma y pueden desencadenar la lisis de las células hospedadoras mediante toxinas, que pueden influir en la adhesión, colonización e invasión, así como la desviación de la respuesta inmune del hospedador62-66. Este importante grupo de proteínas se pierde durante el procesamiento de las bacterias intactas67,68. Las proteínas de las vesículas de membrana externa (OMV) constituyen un subproteoma muy importante en los patógenos, ya que desencadenan reacciones inmunogénicas potentes al ser liberadas por el microorganismo al medio extracelular. Las vesículas producidas por Neisseria meningitidis han sido utilizadas por su efecto inmunoprotector frente a este patógeno69,70.
BiopelículasLas biopelículas están formadas por una biocapa robusta de microorganismos y matriz extracelular. Esta estructura permite a las células que la componen la unión a superficies artificiales o bióticas, dificultando la eliminación de los microorganismos que la componen71. Las biopelículas bacterianas representan una antigua estrategia de supervivencia procariota. Donlan72 definió la biopelícula como una comunidad microbiana sésil, caracterizada por células que están adheridas irreversiblemente a un sustrato o interfase, o unas con otras, embebidas en una matriz de sustancias poliméricas extracelulares que ellas han producido exhibiendo un fenotipo alterado en relación con la tasa de crecimiento y transcripción génica. La biopelícula proporciona a las bacterias embebidas en ella ventajas significantes frente a fluctuaciones medioambientales de humedad, temperatura y pH y reserva de nutrientes73. Por este motivo se estudiaron las diferencias en la abundancia de proteínas entre las células crecidas en biopelícula y células plantónicas mediante la técnica 2DE con tinción de plata en Candida albicans. Como resultado de esta comparación, se observó que las células crecidas en biopelícula presentaban una menor capacidad antioxidativa además de presentar la proteína Pil1p, proteína sensible al tratamiento con un tipo determinado de antifúngicos74. Este resultado permitió encontrar una diana y un tratamiento eficaces contra el estado más resistente de estas levaduras.
Identificación de microorganismos y sensibilidad a antimicrobianosLa identificación de los microorganismos causantes de infección es preciso que se logre en el mínimo tiempo posible. Por ello, un campo amplio de investigación es la búsqueda de técnicas de identificación alternativas a las fenotípicas/metabólicas, que requieren, además del aislamiento del microorganismo, entre 18-24h más para la identificación de este. Una opción para la identificación rápida de microorganismos es la optimización de la MS. En la revisión de Jordana-Lluch et al.75 se explica detalladamente el uso de esta técnica y los avances obtenidos hasta el momento.
La aplicación de la MS en la identificación de microorganismos requiere únicamente del aislamiento y una media de 5min; sin embargo, en este proceso no se obtienen datos sobre la sensibilidad a antimicrobianos. Para obtener dicha información es necesaria la realización de pruebas de sensibilidad del microorganismo, pero dichas pruebas requieren de un tiempo añadido. Se han realizado estudios utilizando la MS para detectar la presencia de betalactamasas, diferenciar entre S.aureus sensibles y resistentes a meticilina, e incluso detectar factores de patogenicidad76-79.
Búsqueda de biomarcadores en fluidos corporalesLos biomarcadores facilitan el desarrollo de herramientas específicas de diagnóstico, monitorizando la respuesta a la terapia o el progreso de la enfermedad80. Cuando analizamos muestras biológicas (de procedencia humana o animal modelo), la dificultad radica en la creación de grupos homogéneos para asegurar una buena correlación de resultados. Por ello es imprescindible la estandarización de la clasificación de las muestras.
La comparativa de fluidos entre pacientes y controles puede permitir la identificación de proteínas diferenciales. Estas proteínas pueden ser utilizadas posteriormente como método de diagnóstico rápido. Un estudio de este tipo se llevó a cabo con el virus de la hepatitisE (HEV), en el que el plasma y la orina de pacientes infectados fueron analizados mediante 2D-DIGE frente muestras control. Se demostró que más de 30 proteínas se expresaban de manera diferencial en pacientes con HEV aguda respecto a los controles81. Estos resultados demuestran la potencialidad de la proteómica en la búsqueda de biomarcadores.
Las proteínas diferenciales obtenidas en la comparación entre pacientes con diferentes grados de la enfermedad podrían ser utilizadas para determinar el pronóstico del paciente.
InmunomaLa inmunoproteómica es una herramienta potencialmente útil en el campo de búsqueda de biomarcadores para enfermedades infecciosas, especialmente en situaciones donde el patógeno es difícil de cultivar in vitro82-85. La tecnología SELDI ha sido aplicada muy recientemente en la identificación de biomarcadores para discernir cepas de Helicobacter pylori asociadas a diferentes estados de la enfermedad86. De esta manera se identifican «firmas» de anticuerpos en suero83,84.
La identificación y caracterización de proteínas antigénicas es un requerimiento para el desarrollo de vacunas, ya que poseen un gran interés como alternativa terapéutica al tratamiento. Para la investigación de la inmunorreactividad de los anticuerpos humanos o anticuerpos de modelos animales frente a las proteínas microbianas, la combinación de 2DE e inmunoblotting representa el método de elección. Extractos proteicos totales del microorganismo patogénico son separados por 2DE y las proteínas inmunorreactivas son detectadas mediante 2D inmunoblots con suero de pacientes infectados, en comparación con el patrón obtenido con sueros control87. Las proteínas que generan señal diferencial entre ambos grupos son identificadas por huella peptídica.
La expresión proteica varía según el estado y condición de crecimiento de los microorganismos. Por esta razón es aconsejable realizar los inmunoblottings con extracto proteico del microorganismo crecido in vivo, para evitar las variaciones transcripcionales, traduccionales y postraduccionales encontradas entre los cultivos in vitro vs in vivo.
La inmunoproteómica es útil para aportar propuestas generales de diagnóstico o para la selección de biomarcadores83. Además, esta metodología puede ser fácilmente aplicada a múltiples infecciones humanas y de veterinaria de gran importancia por su incidencia y severidad88-96.
Vacunas y fármacosLa selección de dianas para fármacos y/o vacunas es esencial para la obtención de un resultado óptimo en el control y/o tratamiento de una enfermedad infecciosa. Los criterios a tener en cuenta para seleccionar una diana son: a)que esta sea esencial para la infectividad y proliferación del patógeno en el hospedador; b)que tenga una baja redundancia en las vías del patógeno, aunque tenga elevada redundancia en las vías del hospedador, siempre y cuando se puedan mitigar los efectos tóxicos; c)que esté amplia y consistentemente expresada en los tejidos de infección y en los diferentes pacientes de la población, y d)que sea terapéuticamente tratable utilizando pequeñas moléculas tales como siRNA, anticuerpos u otras modalidades farmacéuticas.
La vacunología reversa es una técnica utilizada en la búsqueda de nuevas dianas. Esta aproximación permite un cribado a favor de las proteínas con potencial inmunogénico. Los datos obtenidos del proteoma durante la infección pueden ser explotados como base para el desarrollo de nuevas quimioterapias antimicrobianas y vacunas. Una baja proporción de estas proteínas puede ser utilizada como diana para quimioterapia antimicrobiana97 o como antígeno para vacunas protectoras98. Algunos patógenos en los que se ha realizado el estudio del inmunoma son: Chlamydia pneumoniae, Haemophilus influenza, Neisseria meningitidis, Helicobacter pylori, Bacillus anthracis, Streptococcus agalactiae, Mycobacterium tuberculosis y Mycobacterium bovis98-111.
ConclusiónLa proteómica es una herramienta verdaderamente útil en el estudio de las enfermedades infecciosas, proporcionando una visión global. Los análisis proteómicos permiten la realización de un estudio básico de la enfermedad, facilitando la búsqueda de marcadores de infección o virulencia. Posteriormente, los resultados obtenidos tienen una aplicabilidad traslacional dirigida al diagnóstico y pronóstico de la enfermedad, además de permitir el desarrollo de nuevas terapias antimicrobianas y el avance en vacunología.
Las técnicas proteómicas no solo permiten la identificación de nuevos marcadores, sino que pueden ser implantadas como técnicas de diagnóstico rápido, pudiendo dirigir el tratamiento con mayor celeridad evitando los cultivos in vitro, que requieren largos tiempos de incubación, retrasando el diagnóstico, además de generar falsos negativos en aquellos microorganismos viables pero no cultivables.
Esta revisión muestra como la proteómica es una herramienta potente en investigación. A día de hoy, muchos expertos consideran las técnicas en proteómica útiles en el descubrimiento de proteínas de interés clínico, como biomarcadores, pero su aplicabilidad asistencial es baja. El resultado obtenido en los trabajos de investigación basados en proteómica es adaptado a técnicas convencionales como el ELISA, pero estas no se mantienen como herramientas en diagnóstico. Esta revisión pretende mostrar a los expertos del ámbito clínico nuevas técnicas disponibles para resolver las dificultades que se presentan día tras día.
Actualmente, la proteómica es un área valorada por su clara utilidad, aunque aún hay mucho trabajo que realizar en ámbitos como las enfermedades infecciosas. Para mejorar su utilidad en el futuro, debemos concienciar a todo el personal implicado en el ámbito asistencial de su importancia, así como integrar los resultados de los estudios de todas las «ómicas». Este último aspecto es de gran importancia, ya que, por ejemplo, el conocimiento del genoma de un microorganismo es de vital importancia para la interpretación de los resultados proteómicos.
Otro aspecto limitante en la microbiología clínica es el aislamiento del microorganismo para su posterior identificación. Eliminando este paso, tanto la genómica como la proteómica mejorarían su aplicabilidad en la identificación del agente patógeno en una infección. Por ello, estas mejoras harían posible la aceptación de las técnicas proteómicas en el ámbito asistencial para la detección y diagnóstico en enfermedades infecciosas, reduciendo tiempo y costes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

