




Introducción
El síndrome de inmunodeficiencia adquirida es actualmente una de las principales causas de mortalidad en el mundo. El tratamiento antirretroviral reduce la replicación viral y la progresión de la enfermedad, por lo que la mortalidad se ha reducido en los países que disponen de tratamiento. Sin embargo, los fármacos actuales son incapaces de erradicar el virus y el tratamiento se concibe "de por vida". Los tratamientos prolongados y su difícil seguimiento (fallos de adhesión) han puesto de manifiesto problemas inicialmente intuidos como la aparición de resistencias a los fármacos antirretrovirales o efectos adversos inesperados como la toxicidad mitocondrial, la redistribución de la grasa corporal y las alteraciones metabólicas. La prevalencia de resistencia primaria o adquirida a los fármacos disponibles está aumentando. Por ello es imperativo encontrar nuevos fármacos que actúen sobre las dianas ya conocidas (transcriptasa inversa y proteasa), así como otros que actúen sobre nuevas dianas y que ambos tengan una mínima toxicidad para el hospedador1,2.
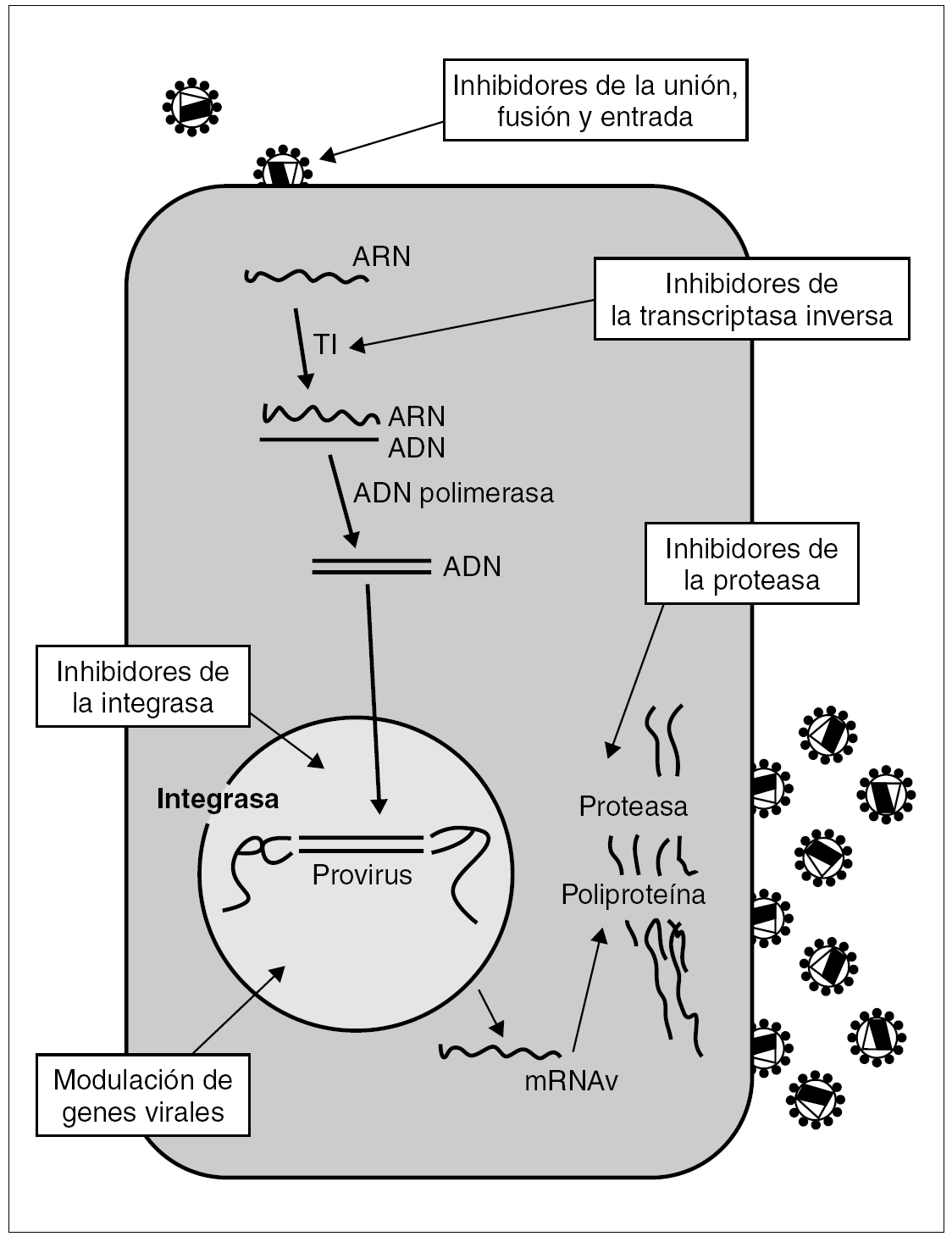
Las dianas terapéuticas más prometedoras son el proceso de unión y entrada del virus a la célula y la integración del ADN viral en el genoma del huésped. A más largo plazo se vislumbran otras posibles dianas como actuaciones sobre el genoma del virus y la posible utilidad de mecanismos biológicos de la célula huésped para inhibir la replicación viral (fig. 1).

Figura 1. Ciclo vital del VIH. Posibles puntos de actuación de los antirretrovirales.
Fármacos que actúan sobre nuevas dianas
Unión y entrada del VIH en la célula
El virus de la inmunodeficiencia humana (VIH) está recubierto por una doble capa lipídica salpicada por glucoproteínas virales entre las que destacan las glucoproteínas gp120 y gp41. Ambas están asociadas y derivan de un precursor común de mayor tamaño: la proteína gp160. La gp120 deriva de la porción aminoterminal de la gp160 y está situada en la parte más externa de la capa lipídica del virus; esta glucoproteína interviene en la unión al receptor CD4 y "dirige" el mecanismo de la fusión. La gp41 deriva de la porción carbono terminal de la gp160; es una proteína transmembrana y tiene un importante papel en la fusión de membranas. La infectividad de un virus requiere la presencia de ambas glucoproteínas3.
La secuencia de aminoácidos de la gp120 está formada por cinco regiones variables (V1 a V5) que alternan con otras más conservadas. Las regiones variables son las más expuestas de la superficie viral. El sitio de unión del virus al receptor CD4 lo forman determinadas regiones de la gp120 que tanto por la secuencia primaria como por su conformación espacial crean un dominio que es capaz de reconocer su diana (receptor CD4) y unirse a la misma con mayor o menor afinidad en función de la existencia o no de mutaciones en la glucoproteína gp120 o en el receptor CD43,4.
Desde el punto de vista fisiopatológico se pueden considerar tres fases o mecanismos en la entrada del virus en la célula4-6 y cada uno de ellos puede ser una diana terapéutica: a) adhesión del virus al receptor CD4; b) unión a los receptores de quimiocinas que actúan como correceptores del VIH, y c) fusión de las membranas celular y viral.
Unión del virus al receptor CD4
Este paso está mediado por la gp120 y el dominio que reconoce el receptor CD4. El aspecto de la gp120 es como una espiga (trímero) formado por las distintas asas (V1-V5) y en la que la región de unión se ha descrito en la parte conservada del tronco de V1-V2 y próximas a V3. Algunas actividades biológicas del virus como tropismo celular, patogenicidad, capacidad de fusión y uso de correceptores parecen estar mediados por un dominio en el asa V3. Tras la unión de gp120 al receptor CD4 se inducen cambios conformacionales que acercan el virus a los receptores de quimiocinas.
Unión a los correceptores
Los cambios en la conformación de la gp120 ponen al descubierto los epítopes que se requieren para la unión a los receptores de quimiocinas. Las quimiocinas son pequeñas proteínas con función proinflamatoria liberadas por macrófagos, linfocitos T activados y otras células mononucleadas (en el caso de MIP-1α, MIP-1β y RANTES) o producidas por células estromales (en el caso de SDF-1) que, cuando se unen a sus receptores, transmiten una señal al interior de la célula activando la quimioatracción. La mayoría de las quimiocinas se pueden agrupar en función de una secuencia de aminoácidos característica en dos familias: CC y CXC. La producción de quimiocinas y la disponibilidad de sus receptores depende de múltiples factores como presencia de citocinas, infecciones por microorganismos, etc.7,8.
Los receptores de quimiocinas utilizados por los retrovirus para entrar en la célula serían los verdaderos receptores del VIH, ya que se ha observado que algunas cepas de VIH modificadas genéticamente que son capaces de entrar en la célula prescindiendo del receptor CD49. El receptor CCR5 de las β -quimiocinas se ha identificado como correceptor de los virus de infección reciente, con tropismo por los macrófagos que tienen este receptor en su membrana y con poca capacidad para formar sincitios en líneas celulares linfoides. Los virus que utilizan este receptor se llaman VIH R5. La importancia del receptor CCR5 en los momentos iniciales de la infección se puede evaluar por el hecho de que las personas que carecen del mismo (homozigotas para la deleción de 32 pares de bases en el gen del CCR5 denominada polimorfismo Δ -32) son resistentes a la infección por el VIH R5 y en los heterozigotos la enfermedad progresa más lentamente. No obstante, estas personas se pueden infectar por vía del correceptor CXCR4, aunque esta eventualidad es muy rara. Por otra parte, los virus que utilizan el correceptor CXCR4 (virus R4) infectan tanto macrófagos como linfocitos; son más habituales en etapas avanzadas de la infección y tienen una elevada capacidad citolítica y formadora de sincitios.
Fusión de las membranas celular y viral
La fusión de ambas membranas es el último paso en el mecanismo de entrada del virus en la célula y está mediada por la gp41. Esta es una glucoproteína transmembrana en la que su porción más distal, aminoterminal, contiene un radical hidrofóbico rico en glicina (donde se formará el péptido de fusión) que es fundamental para la unión de las membranas. Entre los extremos N-terminal y C-terminal de la gp41 existen dos estructuras helicoidales o dominios de repetición heptavalentes (en inglés, heptad repeats, HR) identificadas cada una de ellas como HR-1 y HR-2. La HR-1 queda distal al virus y más próxima al péptido de fusión. En el virión libre la gp41 tiene una conformación no fusogénica, pero cuando la gp120 se une a su receptor, la gp41 sufre un cambio conformacional formándose una elongación (prehorquilla intermedia) que inserta el péptido de fusión en la membrana de la célula diana. Tras la inserción, el HR-2 se pliega en tres helicoides sobre las tres de HR-1 formando una estructura en seis hélices cuyo objetivo es acortar más la distancia entre el virus y la célula hasta poner en contacto ambas membranas que acaban fusionándose.
Bloqueo de la entrada del virus
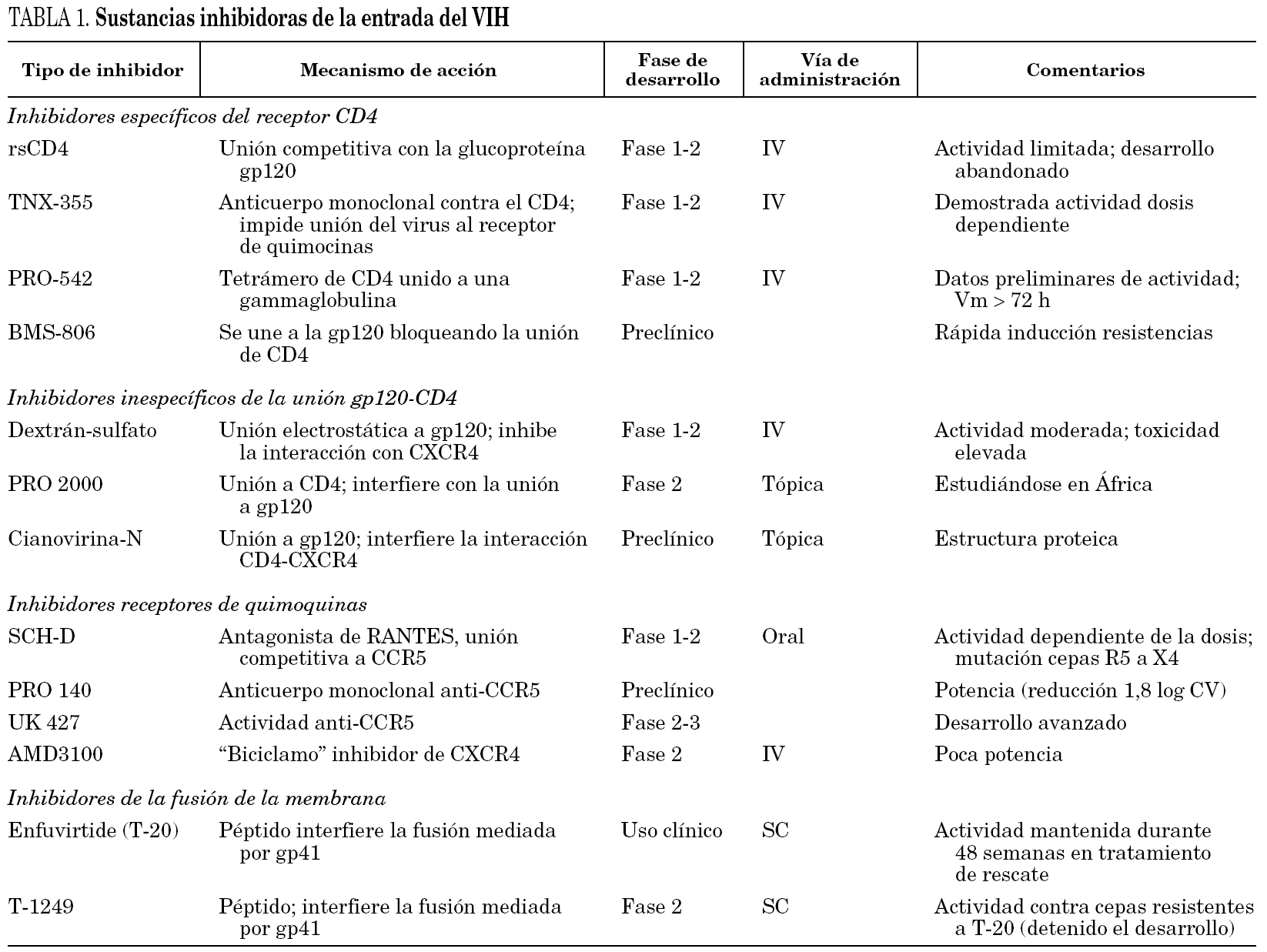
Los fármacos que actúan sobre cualquiera de los tres pasos mencionados pueden bloquear la entrada del virus en la célula. Sus dianas pueden ser cualquiera de las dos glucoproteínas del virus (gp120 y gp41) o sus correspondientes receptores o correceptores celulares. El número de sustancias con esta capacidad que se está estudiando es muy elevado, la mayoría en fases muy precoces de investigación y posiblemente pocas llegarán a ser usadas en la práctica clínica (tabla 1). Sin embargo, ya se dispone de un fármaco (T-20, enfuvirtida o Fuzeon®) cuyo papel como componente de los tratamientos antirretrovirales se está definiendo.

Inhibidores de la unión del VIH al receptor CD4
Inhibidores inespecíficos
Moléculas polianiónicas. Ciertos polisacáridos sulfatados (dextrán-sulfato, pentosán sulfato o heparina) pueden inhibir la replicación viral in vitro. La actividad anti-VIH de compuestos con estructuras tan heterogéneas parece deberse a que poseen una elevada densidad de cargas negativas (polianiones). El dextrán-sulfato se uniría al asa V3 del gp120 de las cepas CXCR4 y evitaría la unión al receptor CD4. Otras sustancias que comparten este mecanismo de acción son el PRO 2000 y la cianovirina N. Aunque su uso en la práctica clínica es improbable, en la actualidad distintos polianiones están siendo evaluados como agentes de uso tópico6.
Inhibidores específicos
CD4 recombinante soluble (rsCD4). El rsCD4 fue uno de los primeros fármacos antirretrovirales estudiado. Actuaría bloqueando la gp120 e in vitro ha demostrado una gran actividad. Sin embargo, su actividad es mucho menor frente a cepas virales obtenidas de enfermos, posiblemente debido a que existen variantes virales de menor afinidad. Por ello, rsCD4 no ha seguido su desarrollo como agente anti-VIH5,6.
Anticuerpos monoclonales anti-CD4. Son anticuerpos monoclonales anti determinados epítopes del receptor CD4 o de la gp120 que al unirse a los mismos bloquean la interacción y reprimen la replicación de numerosos subtipos de virus. Aunque el bloqueo de los receptores CD4 podría causar cierta inmunodepresión, al menos el anticuerpo llamado TNX-355 ha sido bien tolerado y no se ha documentado linfocitopenia CD4+5. Un primer ensayo clínico (fase I/II) a dosis única de TNX-355 demostró una reducción en la carga viral y aumento de células CD4+ a los 21 días10. Sin embargo se requieren ensayos clínicos más prologados que confirmen la utilidad de este anticuerpo. El PRO-542 es un tetrámero híbrido que contiene dominios del receptor de CD4 unidos a una IgG2 y que actúa como un señuelo del receptor CD4 al que se une la gp120 viral, bloqueando su unión al verdadero receptor CD4. In vitro ha demostrado actividad con cepas tanto de laboratorio como clínicas y esta actividad se ha confirmado in vivo en un número reducido de enfermos en estadios avanzados11.
BMS-806. Esta sustancia forma parte de otras moléculas que pueden unirse de modo competitivo y reversible a la gp120 bloqueando la interacción con el receptor CD4. Se ha evaluado in vitro y es activa frente a diferentes cepas de VIH, pero la aparición precoz de resistencias relacionadas con mutaciones en su punto de actuación en la gp120 le hace perder eficacia12,13.
Sustancias que bloquean la unión a los receptores de las quimiocinas
El descubrimiento de los correceptores para la entrada del virus ha abierto nuevas posibilidades para el desarrollo de fármacos anti-VIH. Se ha intentado emular la estructura de las quimiocinas naturales que bloquean estos receptores e inhiben la replicación viral14,15. Se están estudiando en fases precoces fármacos que bloquean uno u otro correceptor, con la duda de si el bloqueo de uno de ellos puede inducir mutaciones en la cepa que facilite que el virus use el otro receptor.
Antagonistas del receptor CCR55
Tak-220. Estudios en fase preclínica de esta molécula han demostrado una alta especificidad para CCR5 sin afinidad por otros ligandos; se ha observado igualmente que con la administración oral se consiguen concentraciones adecuadas del fármaco en sangre.
SCH-C/SCH-D. Se trata de sustancias con gran actividad intrínseca contra cepas R5 y con actividad sinérgica con otros fármacos antirretrovirales. SCH-C ha sido probado en un ensayo fase I/II en 12 enfermos durante 10 días demostrando una reducción de la carga viral entre 0,5 y 1,0 log. Sin embargo, no se ha seguido su desarrollo por riesgo de arritmias cardíacas al documentarse un alargamiento del espacio QTc16,17. Ha sido sustituido por SCH-D, más potente según se ha comunicado recientemente (reducción media de carga viral 1,3 log10), mejor tolerado y no tiene efecto sobre el ritmo cardíaco18. Sin embargo, hay que hacer notar que en uno de los 48 pacientes estudiados desarrolló una cepa viral mixta R5/X4 y en otro apareció una cepa X4 al finalizar el tratamiento.
UK-427,857. Es un fármaco activo contra un gran número de cepas virales y específico para el receptor CCR5 (inactivo frente a cepas CXCR4). Actualmente se están realizando estudios en fase II/III de su desarrollo.
Otras sustancias. Otras sustancias que están en estadios más precoces son el Pro-140 que es un anticuerpo monoclonal específico y potente (reducción de carga viral hasta 1,8 log) que inhibe la entrada del virus y no bloquea la actividad del receptor CCR5. El GW873140 que ha demostrado buena tolerancia (molestias digestivas) y una vida media prolongada tras la administración oral19 y el AMD887 que in vitro ha demostrado actividad y potencia tanto como único fármaco o asociado a AMD070 (inhibidor del correceptor CXCR4)20.
Antagonistas del receptor CXCR45
Biciclamos. Son moléculas que reciben este nombre por estar constituidas por dos anillos macrocíclicos (ciclamos) unidos por una cadena alifática o aromática. Son fármacos muy potentes y activos frente a VIH-1 y VIH-2 en cultivo celular, pero se han descartado como fármacos en ensayos clínicos. AMD3100 es el biciclamo del que se tienen más datos. En estudios in vitro en los que células mononucleadas periféricas infectadas con cepas X4 y R5 se pusieron en contacto con AMD3100, se observó un bloqueo del receptor CXCR4 y sólo se recogieron en el sobrenadante virus R5. En otro estudio con linfocitos de sangre periférica que fueron infectados por virus formadores de sincitios, estas cepas perdieron la capacidad de formar sincitios al mutarse para usar el correceptor CCR521. Esta mutación puede reducir la evolución de la enfermedad. Sin embargo, el desarrollo del AMD3100 se ha detenido por los efectos adversos en un estudio en fase I, pero se están estudiando otros compuestos como el AMD070 que ha demostrado actividad in vitro frente a un gran número de cepas multirresistentes a los fármacos actuales.
El KRH-1636 y KRH-2731 son antagonistas potentes del receptor CXCR4 de administración oral y activo en estudios preclínicos22,23.
Bloqueo de la fusión del virus con la membrana celular
Las sustancias que actualmente se conoce que tienen capacidad para bloquear la fusión de la cápside viral y la membrana celular son péptidos sintéticos que mimetizan la región HR-2 de la gp415. Actúan sobre la horquilla de ataque, inhibiendo su formación. Esta horquilla de ataque es necesaria para la fusión del virus. Entre estas sustancias destaca la enfuvirtida (T-20) y el T-1249.
La enfuvirtida es un péptido de 36 aminoácidos que mimetiza una secuencia de la región HR-2 y es activo frente a cepas X4 y R5. La enfuvirtida se une al dominio HR-1 de la gp41 del virus, evitando que se forme la estructura en seis hélices que se requiere para iniciar los cambios de conformación que acaban en la fusión de membranas. El fármaco es activo durante la fase de la aproximación del virus a la célula diana en la que la gp41 y específicamente el dominio HR-1 es accesible. Esta "ventana terapéutica" podría variar en función de la afinidad del virus por el receptor, de tal modo que si la afinidad es muy elevada el tiempo hasta la acción del fármaco sería más corto y posiblemente su eficacia sería menor en estas cepas virales5. La eficacia de enfuvirtida no se modifica independientemente del correceptor utilizado por el virus24.
La enfuvirtida está comercializada en muchos países. Los estudios iniciales que administraron el fármaco en monoterapia por vía intravenosa demostraron un efecto antirretroviral potente con pocos efectos adversos. Esta actividad se demostró tanto para el subtipo B (dominante en Europa y EE.UU.) como frente a otros subtipos de virus. La dosis óptima por vía subcutánea se determinó inicialmente en bomba de perfusión continua y posteriormente en dos dosis diarias en 16 adultos, que negativizaron la carga viral (< 500 copias/ml) cuando la dosis era superior a 100 mg. Estudios in vitro han demostrado sinergia de enfuvirtida con otros fármacos bloqueantes de la unión como AMD-3100, SCH-C y PRO-54225.
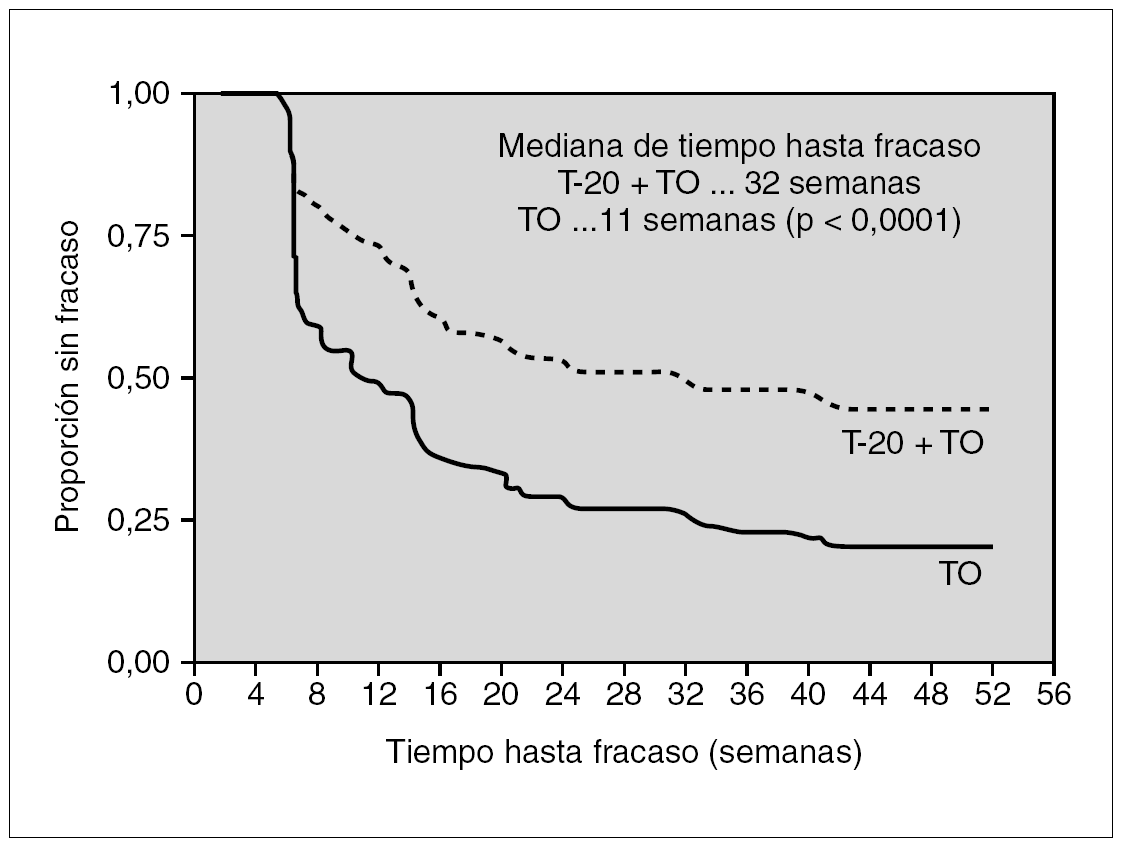
Los resultados de dos estudios paralelos (TORO-1 y TORO-2) en los que se incluyeron 995 enfermos ampliamente pretratados han sido determinantes para que las autoridades sanitarias aprobaran su uso en la práctica clínica. En estos ensayos se comparó la respuesta al mejor tratamiento posible (de 3 a 5 fármacos) determinado mediante estudios de resistencia genotípica y fenotípica añadiendo a un grupo de ellos (proporción 2:1), enfuvirtida. A las 24 y 48 semanas, tanto la reducción de la carga viral como el incremento de los linfocitos CD4+ fueron más elevados en el grupo al que se asoció enfuvirtida. La mediana de tiempo hasta el fracaso virológico (teniendo en cuenta que se trataba de pacientes multitratados) mejoró de 11 a 32 semanas26-28 (fig. 2).

Figura 2. Respuesta de enfermos con resistencia a fármacos de las tres familias a un tratamiento optimizado con o sin enfuvirtida.
Los efectos adversos fueron similares en ambos grupos, salvo las reacciones locales en el punto de inyección. La incidencia global de enfermedades bacterianas fue similar en ambos grupos, pero en el grupo tratado con enfuvirtida se observó una mayor incidencia de neumonías sin que en la actualidad se pueda explicar la causa. En cuanto a alteraciones de laboratorio se documentó un incremento de la eosinofilia periférica en la rama de enfuvirtida, aunque los casos de hipersensibilidad al fármaco son raros. La reacción en el punto de inyección consiste en nódulos inflamatorios dolorosos que ocurren en la mayoría de los enfermos y duran de 1 a 3 días. Solamente una proporción mínima de enfermos alteró sus actividades diarias o requirió analgesia por los nódulos, aunque el 2,8% dejaron el tratamiento por esta causa29.
La eficacia de enfuvirtida en monoterapia es transitoria por lo que se debe asociar a otros antirretrovirales a los que el virus sea susceptible. La resistencia a la enfuvirtida se relaciona con mutaciones en el gen de la gp41. Los dominios HR-1 y HR-2 son relativamente estables, pero los casos documentados de resistencia se relacionan con mutaciones en los codones 36 a 45 de la gp41 con una reducción de la susceptibilidad variable entre 9,1 y 45 veces. Los virus que presentan estas mutaciones tienen una capacidad replicativa (fitness) reducida25.
La enfuvirtida está indicada en pacientes infectados por el VIH en los que la administración de regímenes con al menos un fármaco de los tres grupos de antirretrovirales no ha conseguido respuesta o que hayan desarrollado intolerancia a los tratamientos anteriores. Dado que los estudios TORO demostraron una mejor respuesta virológica si la enfuvirtida se asociaba a fármacos activos, el fármaco se debe administrar antes de que los pacientes hayan agotado las posibilidades terapéuticas.
T-1249. Es un inhibidor de la fusión de segunda generación. Se trata de un péptido de 39 aminoácidos diseñado a partir de diferentes regiones del HR-2 del VIH-1, VIH-2 y virus de la inmunodeficiencia en simios (SIV). Al igual que enfuvirtida, T-1249 muestra una elevada capacidad de reducir la replicación viral en cepas de VIH multirresistentes a los fármacos actuales, incluso resistentes a T-20, y de los subtipos virales A a G. En pacientes no tratados previamente y en monoterapia, T-1949 ha demostrado ser muy potente en una sola dosis diaria (descenso de la carga viral de 2,0 log)5,30. Sin embargo, el programa de desarrollo clínico de este producto se ha detenido temporalmente por dificultades técnicas.
Se están diseñando otros péptidos menos complejos que actuarían en la porción más distal (aminoterminal) de la gp41 próxima al péptido de fusión31, aunque el desafío en el momento actual está en conseguir fármacos que sean capaces de bloquear estos pasos del ciclo del virus, pero que se administren por vía oral de modo que simplifiquen el tratamiento de esta enfermedad crónica.
Integración del ADN proviral (inhibidores de la integrasa)
La integración del ADN proviral en el cromosoma del huésped es un paso necesario en el ciclo replicativo del VIH y la integrasa es la enzima clave de la integración. Esta enzima está codificada en el genoma del VIH junto a la proteasa y la transcriptasa inversa. La integrasa es una diana terapéutica muy atractiva ya que, además de ser esencial en la replicación del virus, no existe en la célula humana una enzima que realice estas funciones, por lo que los fármacos que la bloqueen no deberían afectar otros procesos metabólicos32.
La integrasa es una proteína de 32 kDa que procesa y transporta el ADN del virus al interior del núcleo y cataliza su inserción en el ADN de la célula huésped mediante dos reacciones secuenciales. En primer lugar elimina dos nucleótidos de cada porción 3´ terminal del ADN viral (procesamiento 3´) y en el siguiente integra el ADN viral en el genoma de la célula huésped mediante una transesterificación (transferencia de la cadena de ADN). Tanto la propia integrasa como el llamado complejo de integración (integrasa-ADN viral) serían dianas de actuación farmacológica33.
A pesar de que se conoce la estructura activa de la integrasa desde principios de la década pasada, no existe ningún fármaco disponible en el momento actual. Se conoce una amplia variedad de sustancias químicas con capacidad de bloquear la integrasa in vitro (oligonucleótidos, análogos de la curcumina, compuestos aromáticos polihidroxilados, dicetoácidos, compuestos basados en estructuras afeoil o galloil, hidrazidas y amidas, tetraciclinas, etc.), pero por desgracia esta actividad no la muestran a nivel celular34,35. Sin embargo, recientemente se ha dado un paso más al comunicarse la estructura del complejo "dominio catalítico-inhibidor", lo cual está generando nuevos impulsos a los estudios de algunas de estas estructuras químicas como los dicetoácidos y los ácidos dicafeoil tartáricos36.
Las dos estructuras más estudiadas son V-165, que in vitro ha demostrado ser potente y activa frente a cepas resistentes a inhibidores de la proteasa (IP) o de la transcriptasa inversa y sinérgico con alguno de los antirretrovirales ya comercializados, y el S-1360, dicetoácido en estudios fase 1 con características de eficacia y tolerancia muy similares.
La integrasa puede sufrir mutaciones en su locus activo, pero a pesar de ello la susceptibilidad del virus a los productos actualmente en estudio no parece reducirse37.
Otras posibles dianas
Además de las dianas mencionadas se están vislumbrando otras posibilidades terapéuticas que en el momento actual son más lejanas y se refieren a los mecanismos que condicionan la latencia del virus, la actuación sobre sus genes o los primeros pasos en la síntesis de proteínas virales.
Un factor que parece influir en la eficacia de la replicación viral es la glucoproteína APOBEC3G (apolipoproteína B, enzima productora de ARN mensajero (ARNm), semejante al polipéptido catalítico 3G o CEM15). La APOBEC3G es un factor antirretroviral intracelular innato que es contrarrestado por la proteína del gen vif de los lentivirus y que es específico de especie. APOBEC3G actúa como una citosina desaminasa y en la cadena de ADN viral cambia la citosina por uracilo durante la retrotranscripción, con lo cual provoca una hipermutación que hace inviables a los viriones de la siguiente generación. Sin embargo, el gen vif del VIH-1 es capaz de bloquear el APOBEC3G evitando su incorporación a los viriones hijos y facilitando su degradación en el proteasoma38,39.
Nuevos fármacos con acción sobre dianas conocidas
Con la búsqueda de nuevos fármacos se intenta incrementar su potencia, modificar los perfiles de resistencia, reducir la toxicidad y simplificar las pautas de administración. Con alguna de estas características los fármacos más próximos a incorporarse al arsenal terapéutico son el inhibidor de la transcriptasa inversa análogo de nucleósido (ITIAN) emtricitabina, el no nucleósido (ITINN) capravirina y los IP atazanavir, fosamprenavir y tipranavir.
La emtricitabina es un análogo de la citidina que se administra una vez al día (200 mg). Su perfil de resistencias es similar a lamivudina, pero su potencia es de 4-10 veces mayor por lo que la mutación M184V, que genera resistencia de alto grado, aparece con menor frecuencia. Ya se dispone de éste en algunos países puesto que ha demostrado una eficacia similar o superior a lamivudina y estavudina y dada su mínima toxicidad mitocondrial es un excelente candidato para combinar con tenofovir40.
El amdoxovir es un análogo de guanosina con actividad similar a abacavir, lamivudina o estavudina y efecto antiviral frente al virus de la hepatitis B. Es activo frente a cepas resistentes a zidovudina, lamivudina, didanosina, zalcitabina y abacavir. Actualmente se están efectuando estudios en fase I y II en pacientes pretratados41, aunque su futuro no está claro al haberse detenido temporalmente su desarrollo.
La capravirina es un ITINN potente frente a cepas virales resistentes a otros ITINN. Se requieren al menos dos mutaciones simultáneas (L100I, K103N, 106) para que se desarrolle resistencia. Actualmente se está probando en estudios fase II en pacientes pretratados con ITINN42.
El TMC-125 es un nuevo ITINN, que in vitro muestra equipotencia en VIH wild-type y cepas de VIH multirresistentes a los no análogos (cepas con las mutaciones L100I, K103N, Y181C, Y188L y G190A). En estudios en fase II realizados en pacientes con al menos dos mutaciones de alta resistencia a neviparina y efavirenz se ha observado una respuesta aceptable y buena tolerancia43.
Atazanavir es un IP azapéptido cuya farmacocinética permite la administración una sola vez al día. Además de la monodosis es el IP que menos comprimidos requiere su tratamiento (2 al día o 3 si se administra potenciado con ritonavir). Ha demostrado potencia similar al efavirenz en pacientes no tratados previamente y en enfermos en su primer fracaso a IP se ha mostrado no inferior a lopinavir/ritonavir (en este caso el atazanavir era potenciado con ritonavir) al año de tratamiento, aunque se desconoce el número de mutaciones a IP presentes al inicio del estudio44. Presenta un perfil de seguridad favorable, no alterando los lípidos plasmáticos ni metabolismo hidrocarbonado, pero produce hiperbilirrubinemia indirecta en una proporción del 10-33% de los pacientes. Además el perfil de resistencias de atazanavir (ATV) parece ser totalmente distinto a otros IP. La mutación I50L, específica de resistencia al ATV, es distinta a la I50V que se observa en los pacientes tratados con amprenavir. La mutación I50L reduce sensiblemente la sensibilidad al ATV, pero aumenta la susceptibilidad a otros IP45-47.
El fosamprenavir es un profármaco del amprenavir. Con esta presentación se reduce de un modo importante el número de comprimidos que se van a tomar y, potenciado con ritonavir, se puede administrar una sola vez al día. La eficacia del fármaco se ha demostrado en enfermos no tratados previamente (estudio Neat en dosificación 2 veces al día y estudio Solo administrado una vez al día) en los que se comparó con nelfinavir siendo en ambos estudios la respuesta al fosamprenavir similar a la del nelfinavir. En pacientes pretratados se ha comparado con lopinavir/ritonavir siendo la respuesta medida como proporción de enfermos que consiguen la indetectabilidad de la carga viral o manteniendo la misma indetectable a las 48 semanas (estudio Context) algo inferior al comparador. A pesar de ello, se ha autorizado la comercialización en algunos países48,49.
El tipranavir es un IP no peptídico que ha demostrado actividad in vitro frente a cepas virales con resistencia a los actuales IP50. En enfermos que presentaban su primer fracaso a IP que fueron aleatorizados a recibir dos ITIAN con tipranavir (500 o 1.200 mg)/ritonavir (100 mg) o saquinavir (400 mg)/ritonavir (400 mg) 2 veces al día, a las 16 semanas el descenso de la carga viral (CV) era de 1,4 y 1,8 log. En pacientes en el segundo fracaso a IP tratados con tipranavir (500 o 1.000 mg)/ritonavir (100 mg) cada 12 h el descenso de carga viral a las 48 semanas era de 2,4 log en el grupo con cinco o menos mutaciones de IP y de 2,2 log en el grupo con más de 5 mutaciones50-52.
El TMC-114 es un IP no peptídico en fases precoces de investigación. In vitro ha demostrado una elevada actividad frente a cepas virales con resistencia de alto nivel frente a los IP actuales. En voluntarios sanos se ha demostrado que dosis superiores a 800 mg consiguen concentraciones en sangre equivalentes a la IC50 de cepas multirresistentes. Se puede potenciar farmacocinéticamente con ritonavir. Sus efectos adversos más importantes son gastrointestinales, cefalea o mareos53,54.

