






La glucoproteína P (PGP) es una proteína de membrana, producto del gen MDR-1, que actúa como bomba expulsora de diversos fármacos, entre ellos los inhibidores de proteasa (IP) del virus de la inmunodeficiencia humana (VIH). Numerosos estudios in vitro, en animales y en pacientes, han analizado las relaciones de esta proteína con la farmacocinética y farmacodinamia de los antirretrovirales, con conclusiones dispares. Por otra parte, las publicaciones que analizan la influencia de los polimorfismos de nucleótido único del gen MDR-1, principalmente el C3435T en el exón 26, y el G2677A/G2677T en el exón 21, con las concentraciones plasmáticas de los antirretrovirales, su eficacia y efectos secundarios también demuestran resultados variables que se han tratado de explicar mediante la influencia de otros polimorfismos como el del citocromo p-450.
P-glycoprotein (PGP) is a membrane protein and product of the MDR-1 gene, which acts as an efflux pump for several drugs, such as protease inhibitors (PI) used in HIV. Numerous studies in vitro, in experimental animals, and in patients have analyzed the relationships between PGP and the pharmacokinetic and pharmacodynamic properties of antiretroviral agents, with differing conclusions. In addition, studies focusing on the impact of single nucleotide polymorphisms in the MDR-1 gene, mainly C3435T in exon 26 and G2677A/G2677T in exon 21, on antiretroviral plasma concentrations, efficacy and adverse effects, have reported varying results, which have been attributed to the influence of other polymorphisms, such as cytochrome P450.
La respuesta de los pacientes con infección por el virus de la inmunodeficiencia humana (VIH) al tratamiento antirretroviral de gran actividad (TARGA) es muy variable, por lo que el tratamiento, tras el primer año, fracasa entre el 20 y el 88% de los pacientes1. Los factores que influyen en este fracaso son diversos y se relacionan con el propio virus, con el tratamiento farmacológico o con el paciente, y es la adherencia al tratamiento un factor clave2-4. Aspectos relacionados directamente con el enfermo y de gran importancia son los factores genéticos que determinan la gran variabilidad en la farmacocinética y farmacodinamia de los antirretrovirales5,6. La farmacogenética trata de explicar estas diferencias interindividuales y estudia cómo influyen los polimorfismos genéticos en la respuesta a los diferentes fármacos7,8. En los últimos años sus avances han ayudado a comprender aspectos relacionados con la respuesta a los antirretrovirales, o sus efectos adversos. Ejemplos de ello son las asociaciones de los haplotipos HLA-B*5701, HLA-DR7, y HLA-DQ3 y hsp70-hom con la hipersensibilidad al abacavir, del genotipo 1 de la enzima uridín difosfato glucuronil transferasa con la hiperbilirrubinemia causada por atazanavir, y del HLA-DRB1*01 con la hepatitis y exantema cutáneo originados por nevirapina6.
En el futuro, la farmacogenética se esboza como una herramienta necesaria para crear fármacos a la carta7,9. Una de las proteínas que más ha estudiado esta disciplina es la glucoproteína P (PGP), una proteína de membrana con un papel clave en los procesos de transporte y eliminación de diversos fármacos como los antirretrovirales10-13.
La glucoproteína PLas células del epitelio intestinal, túbulo renal, placenta, testículos y barrera hematoencefálica (BHE) expresan diversos tipos de proteínas transportadoras que sirven para bombear fuera de la célula sustancias potencialmente tóxicas, como, por ejemplo, fármacos11. Una de estas proteínas es la PGP, una proteína transmembrana de 170 kD, compuesta por 1.280 aminoácidos con seis dominios, perteneciente al grupo de las proteínas ABC (ATP-binding cassette). Está compuesta por dos homólogos simétricos que tienen como misión transportar azúcares, péptidos, polisacáridos, nucleósidos, proteínas y algunos fármacos desde el interior al exterior de la célula, obteniendo la energía necesaria para este transporte, contra gradiente de concentración, de la hidrólisis del ATP10-13.
La PGP es codificada por el gen ABCB-1 (ATP-binding cassette, subfamilia B) también llamado gen MDR-1 (Multi Drug Resistance)12. En roedores existen tres genes mdr (mdr-1a, mdr-1b y mdr-2) y en el hombre dos MDR (MDR-1 y MDR-3). Los genes MDR-1, mdr-1a y mdr-1b codifican la PGP, mientras que los genes MDR-3 y mdr-2 codifican una ATPasa12,13.
La PGP se expresa en multitud de tejidos: tejido hematopoyético, células sanguíneas, BHE, hepatocitos, células del túbulo proximal renal, enterocitos, epitelio del plexo coroideo, placenta, ovarios y testículos10. La PGP no es imprescindible para la vida de los ratones como demuestran los estudios con ratones knock-out que tienen una viabilidad y supervivencia normal. Estos mismos estudios han demostrado una cierta especialización: la PGP codificada por el gen mdr-1a se expresa en el hígado y el riñón, mientras que la PGP codificada por el gen mdr-1b se expresa en el intestino, la BHE y la placenta13.
Aunque esta revisión se centra en la importancia de la PGP dentro de la infección por VIH, hay que destacar que buena parte de la literatura médica que se refiere a esta glucoproteína se ha centrado en la relación de la PGP con la respuesta al tratamiento en diversos tumores y enfermedades hematológicas.
PGP y fármacosSon sustratos de la PGP bien documentados fármacos como la digoxina, diversos antitumorales, los anticalcineurínicos (ciclosporina y tacrolimus), algunos antiepilépticos y los inhibidores de la proteasa (IP) del VIH10,11. La PGP actúa sobre ellos con su acción de bomba expulsora con especial repercusión en el intestino, los riñones y la vía biliar10. En el intestino causa una secreción hacia la luz de sus sustratos11,14 como por ejemplo los IP, lo que se manifiesta con una limitación de su absorción15. En el túbulo renal se localiza igualmente en la membrana apical y expulsa hacia la orina los fármacos que acceden a la célula desde la sangre16. También se localiza en la membrana apical del canalículo biliar, donde causa un efecto similar17.
Así pues, tanto por su efecto intestinal, como renal y biliar, la PGP contribuye a una disminución de la absorción y a un aumento de la eliminación del organismo de los fármacos que son sus sustratos. Por ello, los inductores de la PGP como la rifampicina, reducen las concentraciones de IP, contribuyendo de manera muy importante a esta acción el efecto inductor simultáneo de la rifampicina sobre el citocromo P-45018. Por el contrario, la inhibición de la PGP aumenta la absorción de los fármacos. Así, el ritonavir, que inhibe la PGP19, aumenta la concentración plasmática de la digoxina20.
Muchos de estos fármacos que actúan sobre la PGP, como el ritonavir y la rifampicina, son a la vez sustratos del CYP3A4, y muchas células coexpresan ambas proteínas. Por ello, se ha sugerido que la expresión conjunta del CYP3A4 y de la PGP en el hígado o el intestino puede tener un papel sinérgico de protección del organismo frente a agentes exógenos10,11. Además, se debe tener en cuenta que la PGP tiene un importante papel en la modulación de la expresión del CYP3A4, lo que puede complicar la predicción de interacciones de fármacos que sean sustratos de ambas proteínas10.
La PGP tiene un papel clave en la distribución tisular de los fármacos, limitando su paso al compartimiento intracelular y a determinados tejidos, especialmente al sistema nervioso central (SNC), a los testículos y al feto a través de la placenta10,11,15. De hecho, la captación celular de los IP por linfocitos CD4+ está limitada por acción de la PGP21,22. Utilizando modelos experimentales con ratones carentes del gen mdr-1, se ha comprobado que la PGP tiene un papel limitante clave sobre la entrada al SNC de fármacos como ciclosporina, digoxina, vinblastina10 y los IP15,23-25. De manera similar la concentración fetal de los IP aumenta notablemente cuando se bloquea la PGP o en ratones sin el gen mdr-124-26. La inhibición farmacológica de la PGP en pacientes, utilizando ritonavir en dosis bajas, también aumenta la concentración de los IP en semen y líquido cefalorraquídeo27.
Puesto que la PGP limita la entrada de diversos fármacos al organismo y su llegada a determinados tejidos y células, puede ser responsable de la ineficacia de los tratamientos con fármacos que son sus sustratos. De hecho, el papel clave de la PGP en la eliminación de algunos fármacos explica la relación entre su expresión y la respuesta al tratamiento de algunas neoplasias y leucemias28. De manera similar la PGP puede contribuir a la limitación del acceso de los fármacos antirretrovirales a santuarios, que actúan como reservorios de virus, en los que el VIH se replica a pesar del TARGA. Éste es uno de los aspectos clave que puede impedir la erradicación del VIH de los pacientes infectados29-31. La PGP se plantea como una atractiva diana terapéutica para facilitar la llegada de los fármacos a estos reservorios, aunque los inhibidores de la PGP pueden actuar sobre otros fármacos administrados simultáneamente que también sean sustratos de la PGP, originando un aumento de su absorción y entrada en distintos compartimientos que conlleve mayor toxicidad.
Polimorfismo de la PGPLa PGP presenta una gran variabilidad en su expresión entre individuos. Esta gran variabilidad interindividual en la expresión de la PGP se debe, al menos en parte, a la presencia de polimorfismos genéticos10,17. Hasta el momento, se han descrito 29 polimorfismos de un sólo nucleótido en el gen MDR-110. De ellos, 26 no alteran la expresión ni la función de la PGP y sólo tres polimorfismos (el C3435T en el exón 26, el G2677A/G2677T en el exón 21 y el T129C en el exón 1b) se han asociado con variaciones en la expresión y/o función de la PGP10,14,32-34. En el polimorfismo C3435T, los homocigotos CC expresan más PGP en el intestino, placenta y en los linfocitos que los TT14,33,34. También se ha descrito que el polimorfismo C3435T puede coexistir con el polimorfismo G2677T/A, e incluso con el polimorfismo C1236T (polimorfismo sinónimo en el exón 12), lo que hace que algunos efectos atribuidos al polimorfismo C3435T puedan deberse realmente a otros10. Los polimorfismos se distribuyen de manera diferente según las poblaciones. Así por ejemplo, el alelo T del polimorfismo C3435T es raro en la población africana y frecuente en varias poblaciones caucásicas y asiáticas8,32. En nuestro país, la distribución de frecuencias del polimorfismo C3435T es similar a la de otros países caucásicos35.
Algunos datos sugieren que el polimorfismo TT, que implica una menor expresión de la PGP, se asocia con una mejor respuesta a fármacos antineoplásicos28, antiepilépticos36, esteroides en niños trasplantados de corazón37 e inhibidores de proteasa en pacientes con infección por VIH38,39. Este efecto beneficioso se suele explicar a través de los efectos sobre la biodisponibilidad y distribución tisular de los fármacos que suponen las alteraciones de la PGP, pero en el caso de los antirretrovirales podría ser contrarrestado por otros factores celulares como el aumento de trifosfatos endógenos que compiten con los análogos de los nucleósidos o la disminución de la timidincinasa que se observa en tratamientos prolongados con zidovudina. Esta disminución de la timidincinasa puede ser secundaria a una sobreexpresión de PGP, que impediría la acumulación intracelular de la cinasa40.
Relaciones entre la PGP y la infección por el VIHLas interacciones de la PGP, el VIH y los antirretrovirales son complejas. Numerosos datos indican que la infección por el VIH y la PGP influyen mutuamente uno sobre otro, bien directamente o como consecuencia de las interacciones, también bidireccionales, entre la PGP y el tratamiento antirretroviral (fig. 1). Estas interacciones incluyen: a) la influencia de la PGP en la infectividad del VIH y progresión de la enfermedad; b) una acción del propio VIH sobre la expresión y función de la PGP; c) acción de los antirretrovirales sobre la expresión y la función de la PGP, y d) influencia de la PGP sobre la farmacocinética y farmacodinamia de algunos antirretrovirales.
 y la infección por el virus de la inmunodeficiencia humana (VIH) son complejas. La PGP parece influir en la infectividad por el VIH y, a su vez, la infección por el VIH altera la expresión de la PGP (a). Por otra parte, algunos antirretrovirales afectan a la expresión de la PGP y, a su vez, la PGP influye en su farmacocinética (b) y farmacodinamia (c).")
Las interacciones entre la glucoproteína P (PGP) y la infección por el virus de la inmunodeficiencia humana (VIH) son complejas. La PGP parece influir en la infectividad por el VIH y, a su vez, la infección por el VIH altera la expresión de la PGP (a). Por otra parte, algunos antirretrovirales afectan a la expresión de la PGP y, a su vez, la PGP influye en su farmacocinética (b) y farmacodinamia (c).
Todo ello hace difícil interpretar las alteraciones de la PGP en pacientes con infección por el VIH tratados con antirretrovirales. Asimismo, es difícil tratar de correlacionar el polimorfismo, la expresión y la función de la PGP con la evolución de la infección en pacientes con infección por VIH con tratamiento antirretroviral.
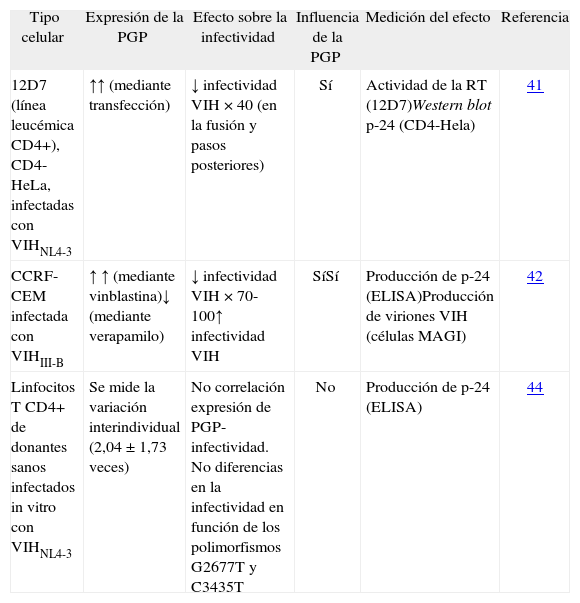
Influencia de la PGP sobre la infección por el VIHEstudios in vitro. Cuando se produce una sobreexpresión in vitro de la PGP en líneas celulares CD4+, se consigue una disminución de la capacidad infectante y de replicación del virus de hasta 100 veces (tabla 1). Esta disminución se debe a una limitación en la fusión del VIH con la membrana celular y también a alteraciones en fases posteriores del ciclo viral, que revierte al inhibir específicamente la PGP con verapamilo41,42. Se han obtenido resultados similares con otros virus como el de la gripe43. Sin embargo, ni las diferencias de dos a tres veces en la expresión de la PGP que se observan fisiológicamente en los linfocitos CD4, ni los diferentes alelos C3435T y G2677A/T se asocian a diferente permisividad de la infección por VIH44.
Modelos in vitro que valoran el efecto de la expresión de la PGP en la infectividad del virus de la inmunodeficiencia humana
| Tipo celular | Expresión de la PGP | Efecto sobre la infectividad | Influencia de la PGP | Medición del efecto | Referencia |
| 12D7 (línea leucémica CD4+), CD4-HeLa, infectadas con VIHNL4-3 | ↑↑ (mediante transfección) | ↓ infectividad VIH × 40 (en la fusión y pasos posteriores) | Sí | Actividad de la RT (12D7)Western blot p-24 (CD4-Hela) | 41 |
| CCRF-CEM infectada con VIHIII-B | ↑ ↑ (mediante vinblastina)↓ (mediante verapamilo) | ↓ infectividad VIH × 70-100↑ infectividad VIH | SíSí | Producción de p-24 (ELISA)Producción de viriones VIH (células MAGI) | 42 |
| Linfocitos T CD4+ de donantes sanos infectados in vitro con VIHNL4-3 | Se mide la variación interindividual (2,04 ± 1,73 veces) | No correlación expresión de PGP-infectividad. No diferencias en la infectividad en función de los polimorfismos G2677T y C3435T | No | Producción de p-24 (ELISA) | 44 |
PGP: glucoproteína P; VIH: virus de la inmunodeficiencia humana.
La relación entre sobreexpresión de PGP inducida in vitro y la disminución de la infectividad del VIH no parece tener relación con el receptor CD4 ni con el correceptor CXCR4 ni CCR5, necesarios para que la infección por el VIH se produzca, puesto que en líneas celulares la sobreexpresión de PGP no se acompaña de una alteración de la expresión de ambas moléculas41,45. Parece que el efecto de la PGP sobre la infectividad y capacidad de replicación del VIH puede deberse a una asociación entre la PGP y unos dominios de membrana ricos en glucoproteínas que tienen gran importancia en la fusión del VIH46.
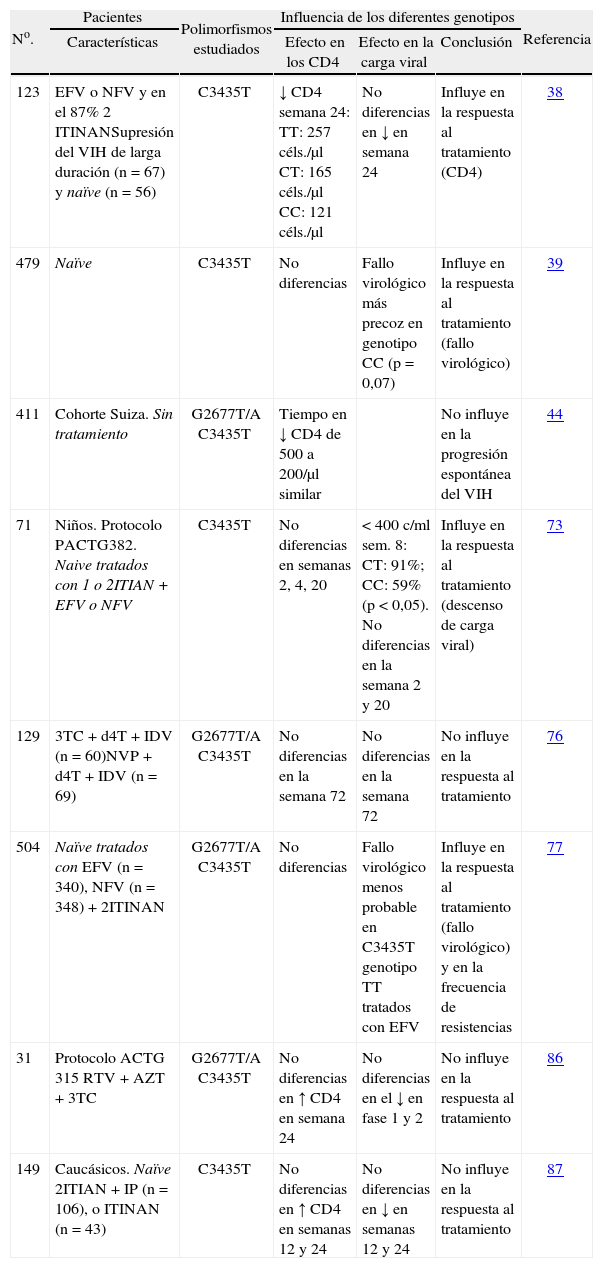
Estudios en humanos. Se ha estudiado la relación de los diferentes polimorfismos del gen MDR-1 con la infectividad de VIH y progresión de la infección por este virus con o sin tratamiento antirretroviral (tabla 2). A pesar de que los resultados de los experimentos in vitro indican una asociación entre la sobreexpresión de la PGP y una menor infectividad del VIH, un estudio prospectivo en 137 pacientes con prácticas de riesgo para adquisición del VIH no encontró diferencias en la distribución de frecuencias de las variantes del MDR-1, T-129C, G2677T/A o C3435T entre pacientes VIH positivos y VIH negativos47.
Efecto del polimorfismo del gen MDR-1 sobre la evolución de la infección por VIH. Estudios en pacientes
| No. | Pacientes | Polimorfismos estudiados | Influencia de los diferentes genotipos | Referencia | ||
| Características | Efecto en los CD4 | Efecto en la carga viral | Conclusión | |||
| 123 | EFV o NFV y en el 87% 2 ITINANSupresión del VIH de larga duración (n = 67) y naïve (n = 56) | C3435T | ↓ CD4 semana 24: TT: 257 céls./μl CT: 165 céls./μl CC: 121 céls./μl | No diferencias en ↓ en semana 24 | Influye en la respuesta al tratamiento (CD4) | 38 |
| 479 | Naïve | C3435T | No diferencias | Fallo virológico más precoz en genotipo CC (p = 0,07) | Influye en la respuesta al tratamiento (fallo virológico) | 39 |
| 411 | Cohorte Suiza. Sin tratamiento | G2677T/A C3435T | Tiempo en ↓ CD4 de 500 a 200/μl similar | No influye en la progresión espontánea del VIH | 44 | |
| 71 | Niños. Protocolo PACTG382. Naive tratados con 1 o 2ITIAN + EFV o NFV | C3435T | No diferencias en semanas 2, 4, 20 | < 400 c/ml sem. 8: CT: 91%; CC: 59% (p < 0,05). No diferencias en la semana 2 y 20 | Influye en la respuesta al tratamiento (descenso de carga viral) | 73 |
| 129 | 3TC + d4T + IDV (n = 60)NVP + d4T + IDV (n = 69) | G2677T/A C3435T | No diferencias en la semana 72 | No diferencias en la semana 72 | No influye en la respuesta al tratamiento | 76 |
| 504 | Naïve tratados con EFV (n = 340), NFV (n = 348) + 2ITINAN | G2677T/A C3435T | No diferencias | Fallo virológico menos probable en C3435T genotipo TT tratados con EFV | Influye en la respuesta al tratamiento (fallo virológico) y en la frecuencia de resistencias | 77 |
| 31 | Protocolo ACTG 315 RTV + AZT + 3TC | G2677T/A C3435T | No diferencias en ↑ CD4 en semana 24 | No diferencias en el ↓ en fase 1 y 2 | No influye en la respuesta al tratamiento | 86 |
| 149 | Caucásicos. Naïve 2ITIAN + IP (n = 106), o ITINAN (n = 43) | C3435T | No diferencias en ↑ CD4 en semanas 12 y 24 | No diferencias en ↓ en semanas 12 y 24 | No influye en la respuesta al tratamiento | 87 |
3TC: lamivudina; AZT: zidovudina; d4T: estavudina; EFV: efavirenz; IDV: indinavir; IP: inhibidores de la proteasa; ITIAN: inhibidores de la transcriptasa inversa análogos de nucleósido; ITINAN: inhibidores de la transcriptasa inversa no análogos de nucleósido; NFV: nelfinavir; NVP: nevirapina; RTV: ritonavir; sem: semana; VIH: virus de la inmunodeficiencia humana.
Respecto a la relación entre polimorfismo del MDR-1 y progresión de la infección por VIH sin tratamiento, el único estudio que valora este aspecto encontró que la media de tiempo que pasaba desde que los pacientes tenían 500 CD4/μl hasta que llegaban a 200 CD4/μl era 3,68 años sin que hubiera diferencias significativas entre los pacientes que tenían los diferentes genotipos C3435T y G2677A/T44.
Por otra parte, varios estudios han analizado la relación entre el polimorfismo del MDR-1 y la respuesta al tratamiento antirretroviral con resultados dispares que se comentarán más adelante (tabla 2).
Influencia de la infección por VIH sobre la PGPEstudios in vitro. En líneas celulares U937 y H9 la infección por VIH originó un incremento de expresión de la PGP de unas 10 veces48. Recientemente, se ha demostrado que la proteína Tat del VIH induce in vitro la expresión de la PGP en líneas celulares de endotelio microvascular cerebral49.
Estudios en animales. En la superficie de las células mononucleares de sangre periférica y de tejido linfoide de monos infectados por el virus de la inmunodeficiencia simio-humana (SVIH), la PGP se expresa menos que en monos no infectados; estos datos son contrarios a los obtenidos in vitro50.
Estudios en humanos. Varios estudios señalan que los linfocitos de pacientes con infección por VIH de sangre periférica de diferentes subtipos (CD40+, CD8+, CD16+, CD19+) muestran una menor expresión y función de la PGP que los de los controles sanos, por lo que se ha llegado a sugerir que la expresión de la PGP podría ser un marcador de la evolución de la enfermedad51-56 (tabla 3). No todos los estudios presentan resultados uniformes; en el estudio realizado por Andreana et al51 se detectó un déficit funcional de la PGP acorde con el resto de trabajos, pero con un aumento de la expresión de la PGP, y en el realizado por Bossi et al57 no se observaron diferencias en la expresión de la PGP de los linfocitos CD4+ de pacientes infectados con VIH respecto a otros no infectados, ni se vio una relación con la carga viral. También se ha estudiado la expresión de la PGP en monocitos de sangre periférica de pacientes VIH, sin encontrar alteraciones respecto al grupo de controles sanos58.
Estudios con pacientes en los que se analiza el efecto de la infección por virus de la inmunodeficiencia humana y el tratamiento antirretroviral sobre la expresión y función de la proteína P
| No. | Pacientes | Efecto sobre la expresión de la PGP* | Efecto sobre la función de la PGP* | Método de medición del efecto | Referencia |
| Características de los sujetos | |||||
| 16 | Grupo I CD4 < 200/μl ± AZT (n = 8)Grupo II CD4 > 200/μl ± AZT (n = 8)Controles (n = 8) | ↑ en CD4+: controles < grupo I < grupo IITratamiento con AZT sin efecto | ↓ en CD4+, CD8+: sanos < grupo I < grupo II | *Citometría con Ac-Mo MRK-16**Acumulación intracelular de Rh 123 | 51 |
| 44 | Estadio A2 (n = 13), B2 (n = 15), C3 (n = 16), 21 con AZT (6 controles sanos) | ↓ en linfocitos CD4+, y CD19+Tratamiento con AZT sin efecto | ↓ en CD16+, CD19+ | *Citometría con Ac-Mo MRK-16**Acumulación intracelular de Rh 123 | 52 |
| 21 | Naïve (n = 4), TARGA (n = 17) (21 controles sanos) | ↓ en linfocitos totalesSin relación con CD4 totales | – | *Citometría con Ac-Mo UIC2 | 53 |
| 38 | 11 asintomáticos 8 LGP* 19 estadio C3 | ↓ en CD8+, CD16+ | – | *Citometría con Ac-Mo MRK-16, y MM4.17 | 54 |
| 18 | 18 con 2 ITIAN +IP, 8 con fallo virológico (21 controles sanos) | En CD4+: sin relación con infección por VIH, fracaso, ni TARGA | Sin relación con infección por VIH, ni TARGA | *Citometría con Ac-Mo MRK-16**Acumulación intracelular de Rh 123 | 55 |
| 32 | 16 asintomáticos, 16 sida, 16 naïve, 16 TARGA | Sin relación con infección por VIH, carga viral o TARGA | – | *Citometría con Ac-Mo MRK-16 | 58 |
| 70 | 35 estadio A, 25 C con TARGA, 20 VIH sin tratamiento | – | ↓ en CD4+ y CD8 en VIH naïve↓ en linfocitos de pacientes con IP: RTV > SQV > NFV > IDV | **Acumulación intracelular de Rh 123 | 65 |
| 185 | 131 con TARGA | – | Mayor actividad en CD4+ asociadas con menor carga viral. Sin relación con el tratamiento | *Citometría de flujo con DiOC2(3) | 72 |
AZT: zidovudina; IDV: indinavir; ITIAN: inhibidores de la transcriptasa inversa análogos de nucleósido. IP: inhibidor de proteasa; LGP: linfadenopatía generalizada persistente; NFV: nelfinavir; RTV: ritonavir; SQV: saquinavir; TARGA: tratamiento antirretroviral de gran actividad; VIH: virus de la inmunodeficiencia humana.
Los IP como amprenavir, atazanavir, fosamprenavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir y tipranavir son sustratos de la PGP10,19,21,22, por lo que si causan alteraciones de la PGP pueden repercutir sobre su propia farmacocinética. Por el contrario, los inhibidores de la transcriptasa inversa no análogos de nucleósidos como efavirenz y nevirapina (ITINAN) no son considerados sustrato de la PGP59, aunque este dato está en duda últimamente, al menos en el caso de la nevirapina60.
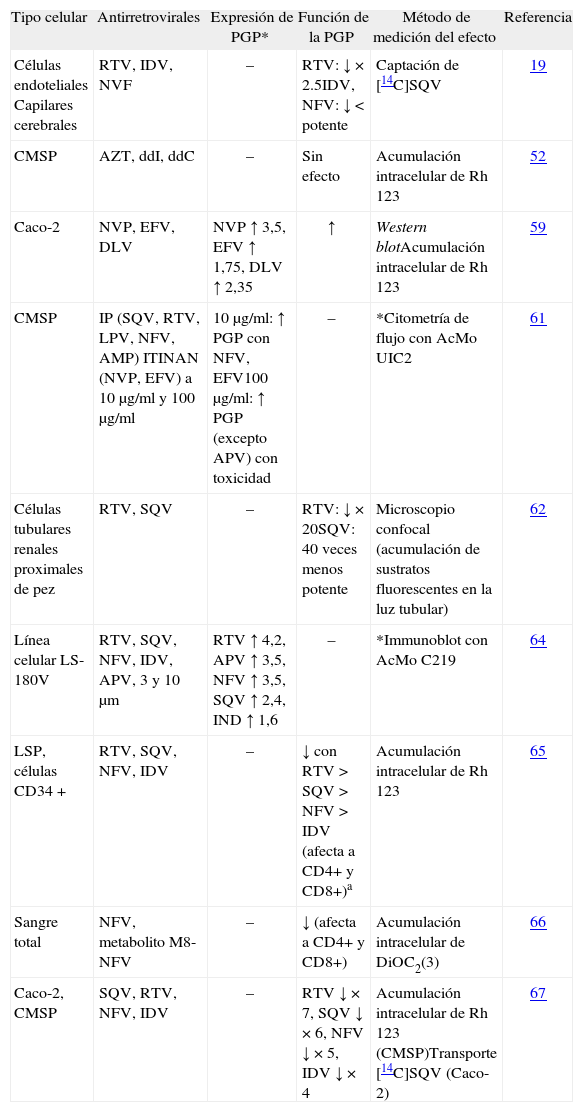
Estudios in vitro. Los inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN) (se han estudiado AZT, DDI y DDC) no parecen tener efectos en la actividad de la PGP en las distintas subpoblaciones linfocitarias de sangre periférica52,58 (tabla 4). Los ITINAN (delavirdina, efavirenz y nevirapina) aumentan la expresión y función de la PGP tanto en líneas celulares como en células mononucleares de sangre periférica59,61. Sin embargo, entre los distintos IP existen diferencias con respecto a los efectos que se producen sobre la expresión y la función de la PGP; el nelfinavir aumenta la expresión de la PGP61, mientras que el indinavir y el ritonavir no la modifican en células mononucleares de sangre periférica58. Respecto a la función de la PGP, todos los IP la inhiben19,24,61-70. En el estudio realizado por Saitoch et al71, el plasma de pacientes tratados con nelfinavir inhibió la PGP de los linfocitos de pacientes no infectados ni tratados, siendo este efecto concentración-dependiente, mayor para el nelfinavir que para su metabolito M8 y mayor sobre CD4+ que sobre CD8+. No obstante, las consecuencias de la inhibición de la PGP que producen los IP en los linfocitos no se conocen bien. Cabe suponer que pueden aumentar la concentración de los antirretrovirales que sean sustrato de la PGP en los linfocitos y, por tanto, la eficacia del tratamiento. Sin embargo, también podrían aumentar la timidincinasa, que es un sustrato de la PGP y, como consecuencia, facilitar la replicación vírica. Por otra parte, los efectos que los antirretrovirales causan sobre la PGP en linfocitos no parecen extrapolables a otras células. Así, por ejemplo, en el estudio de Lucia et al58, la expresión de la PGP en monocitos de sangre periférica procedente de pacientes con infección VIH no se vio afectada por el tratamiento antirretroviral.
Efecto de la exposición a antirretrovirales en la expresión o función de la PGP. Modelos in vitro
| Tipo celular | Antirretrovirales | Expresión de PGP* | Función de la PGP | Método de medición del efecto | Referencia |
| Células endoteliales Capilares cerebrales | RTV, IDV, NVF | – | RTV: ↓ × 2.5IDV, NFV: ↓ < potente | Captación de [14C]SQV | 19 |
| CMSP | AZT, ddI, ddC | – | Sin efecto | Acumulación intracelular de Rh 123 | 52 |
| Caco-2 | NVP, EFV, DLV | NVP ↑ 3,5, EFV ↑ 1,75, DLV ↑ 2,35 | ↑ | Western blotAcumulación intracelular de Rh 123 | 59 |
| CMSP | IP (SQV, RTV, LPV, NFV, AMP) ITINAN (NVP, EFV) a 10 μg/ml y 100 μg/ml | 10 μg/ml: ↑ PGP con NFV, EFV100 μg/ml: ↑ PGP (excepto APV) con toxicidad | – | *Citometría de flujo con AcMo UIC2 | 61 |
| Células tubulares renales proximales de pez | RTV, SQV | – | RTV: ↓ × 20SQV: 40 veces menos potente | Microscopio confocal (acumulación de sustratos fluorescentes en la luz tubular) | 62 |
| Línea celular LS-180V | RTV, SQV, NFV, IDV, APV, 3 y 10 μm | RTV ↑ 4,2, APV ↑ 3,5, NFV ↑ 3,5, SQV ↑ 2,4, IND ↑ 1,6 | – | *Immunoblot con AcMo C219 | 64 |
| LSP, células CD34 + | RTV, SQV, NFV, IDV | – | ↓ con RTV > SQV > NFV > IDV (afecta a CD4+ y CD8+)a | Acumulación intracelular de Rh 123 | 65 |
| Sangre total | NFV, metabolito M8-NFV | – | ↓ (afecta a CD4+ y CD8+) | Acumulación intracelular de DiOC2(3) | 66 |
| Caco-2, CMSP | SQV, RTV, NFV, IDV | – | RTV ↓ × 7, SQV ↓ × 6, NFV ↓ × 5, IDV ↓ × 4 | Acumulación intracelular de Rh 123 (CMSP)Transporte [14C]SQV (Caco-2) | 67 |
APV: amprenavir; AZT: zidovudina; CMSP: células mononucleares de sangre periférica; ddC: zalcitabina; ddI: didanosina; DLV: delavirdina; EFV: efavirenz; IDV: indinavir; IP: inhibidor de proteasa; ITINAN: inhibidores de la transcriptasa inversa no análogos de nucleósido; LPV: lopinavir; LSP: linfocitos de sangre periférica; NFV: nelfinavir; NVP: nevirapina; RTV: ritonavir; SQV: saquinavir.
Estudios en animales. Las alteraciones en la expresión de la PGP detectadas en algunos estudios in vitro no implican una alteración funcional en la PGP en modelos animales. Por ejemplo, el ritonavir (que inhibe la PGP) en dosis altas no es capaz de aumentar el acceso del saquinavir (un sustrato de la PGP) al SNC en roedores24 y el efavirenz no altera la expresión de PGP en el intestino de ratón72. En monos infectados por VIHS se ha estudiado la expresión de la PGP en la superficie de las células mononucleares de sangre periférica y de tejido linfoide demostrando que en los tratados con AZT, 3TC e indinavir se expresa menos PGP que en los tratados con placebo50.
Estudios en humanos. El tratamiento con AZT no altera la expresión ni la función de la PGP en linfocitos totales de sangre periférica ni en subpoblaciones CD4, CD8, CD16 o CD19 positivas de pacientes con infección por el VIH52. En un estudio no se detectaron diferencias en la expresión ni función de la PGP de los linfocitos CD4 entre tratados y no tratados con IP57. En otro tampoco se observó una influencia significativa del tratamiento respecto a la actividad de la PGP en linfocitos CD4 y CD8 de sangre periférica73.
Glucoproteína P y respuesta al tratamiento de la infección VIHPGP y concentraciones plasmáticas de antirretroviralesAunque algunos estudios indican una relación entre el polimorfismo de la PGP y las concentraciones plasmáticas de algunos antirretrovirales, estos hallazgos se deben interpretar con precaución, ya que podrían depender de la ingesta de componentes de la dieta u otros factores ambientales que afecten a la PGP y que tengan efectos inductores o inhibidores del metabolismo a través del CYP3A410.
Estudios en animales. La importancia de la PGP como limitadora de la absorción de los IP y favorecedora de su eliminación se demuestra en ratones sin gen mdr-1a, en los que la concentración de saquinavir, nelfinavir o indinavir tras su administración oral es de dos a cuatro veces mayor que en ratones normales15,24. Por otra parte, los inhibidores de la PGP aumentan las concentraciones plasmáticas de IP en animales24.
Estudios en humanos. Los principales estudios que han analizado la relación entre polimorfismo del gen MDR-1 y concentraciones de antirretrovirales muestran resultados variables. En contra de lo esperable, se ha descrito que los pacientes con genotipo 3435CC (asociado a alta expresión de PGP) tenían mayores concentraciones plasmáticas de efavirenz y de nelfinavir que los pacientes con genotipo TT38. Por el contrario, en otro estudio, los pacientes con genotipo 3435CC tenían menores concentraciones plasmáticas de nelfinavir71 y menores concentraciones valle de nevirapina74. Otros estudios no han encontrado diferencias en las concentraciones plasmáticas de efavirenz75,76, lopinavir75, indinavir76 y nelfinavir77 en los pacientes con genotipo 3435CC frente a 3435TT. El polimorfismo del exón 21 (G2677A/T) tampoco se ha asociado a diferencias en las concentraciones plasmáticas de efavirenz o nelfinavir77.
PGP y concentraciones tisulares de antirretroviralesEstudios in vitro. La entrada dentro de la célula de los IP viene determinada por la actividad de la PGP como se pone de manifiesto en experimentos in vitro en los que se detecta una disminución drástica de la concentración intracelular de nelfinavir, indinavir y ritonavir en las líneas celulares CEMVBL, que hiperexpresan PGP, respecto a las líneas CEM que no la hiperexpresan22,23. También se ha comprobado una gran diferencia en la concentración celular de saquinavir entre líneas celulares que expresan PGP (línea celular Dx5) y células que no la expresan (línea MES-SA)25. Además, líneas celulares epiteliales de riñón transfectadas con el gen MDR-1 transportan muy eficazmente ritonavir y saquinavir24. Por otra parte, las células que hiperexpresan PGP son menos susceptibles a la acción antiviral del AZT, debido a que se alcanzan concentraciones intracelulares menores78.
Sin embargo, la expresión de la PGP varía de forma importante de una células a otras y, por tanto, no pueden extrapolarse los datos obtenidos de un tejido a otro79. De especial relevancia en la infección por VIH puede ser la expresión de PGP linfocitaria y monocitaria. Los estudios indican que su expresión es mayor en monocitos que en linfocitos51,52.
El acceso al espacio intracelular está limitado, además de por la PGP, por otras proteínas de membrana como la asociada a multirresistencia a fármacos (MRP) y la proteína de resistencia del cáncer de mama (BCPR)11,21,40,42. In vitro la inhibición de cada una de estas proteínas supone un aumento de las concentraciones intracelulares de saquinavir80.
Estudios en animales. Los estudios en animales confirman de manera consistente el papel de la PGP como limitador del acceso de los IP a los tejidos. El LY-335979, que es un potente inhibidor de la PGP, aumentó las concentraciones de nelfinavir en testículos y cerebro de ratones 4 y 15 veces, respectivamente, en comparación con la concentración plasmática23. En modelos con ratones deficientes del gen mdr-1, se observó una acumulación de IP en cerebro y testículos con un incremento del cociente concentración tisular/plasmática de 2 a 10 veces15,25. También aumenta la concentración fetal de saquinavir en ratones deficientes de los genes mdr-1a y mdr-1b, y en ratones no deficientes cuando se tratan con inhibidores de la PGP como PSC833 o GG91826.
Estudios en pacientes. Aunque en un estudio con pacientes tratados con antirretrovirales se ha correlacionado de manera inversa la expresión del gen MDR-1 (ARNm) con la concentración intracelular de varios IP (nelfinavir, indinavir, amprenavir, ritonavir)81, otros no han encontrado correlación entre la expresión de la PGP en la superficie de células mononucleares de sangre periférica medida con citometría de flujo y la concentración intracelular de nelfinavir82, saquinavir o ritonavir83 en pacientes tratados con estos fármacos. A pesar de ello, la asociación de ritonavir, que inhibe la PGP, con el tratamiento aumenta el cociente concentración intracelular/plasmática de otros IP81,82. En otro estudio en el que se administró a pacientes una dosis baja de ritonavir (100 mg /12 h) aumentó la concentración de indinavir en suero, en semen y en LCR, y el aumento intracelular resultó mayor de lo esperado para el aumento observado en la concentración sérica, lo que se atribuyó a la inhibición de la PGP27. Las concentraciones de efavirenz dentro de células mononucleares de sangre periférica en pacientes tratados con este fármaco no tienen relación con la expresión de la PGP84. Sin embargo, la concentración intracelular de nevirapina se correlaciona inversamente con la expresión de la PGP85.
PGP y respuesta al tratamiento antirretroviralLos resultados de los estudios que analizan la influencia de los polimorfismos del gen MDR-1 en la respuesta al tratamiento antirretroviral no son uniformes (tabla 2). En 123 pacientes con infección por el VIH se encontró una respuesta al tratamiento antirretroviral, medida como ascenso de CD4 a los 3 y 6 meses de iniciar el tratamiento, significativamente mayor en los pacientes con genotipo 3435TT, sin encontrar diferencias en la caída de la carga viral entre los genotipos TT, CT y CC38. En 479 pacientes de nuevo diagnóstico, los pacientes con genotipo 3435CC presentaron menor tiempo hasta el fracaso virológico (definido por una carga viral > 500 copias/ml) que los genotipos 3435CT y TT, aunque con diferencias en el límite de la significación estadística39. Este estudio no encontró diferencias entre los diferentes genotipos en el desarrollo de resistencias a antirretrovirales. En niños, el estudio PACTG382 encontró una menor proporción de niños que alcanzaban carga viral inferior a 400 copias/ml en la semana 8 de tratamiento antirretroviral en los de genotipo 3435CC (59%) que en los de genotipo 3435CT (91%)73. Un dato discordante en este estudio fue la respuesta de sólo el 57% en los niños con genotipo 3435TT, que los autores consideraron no valorable debido al escaso número de pacientes de este grupo (sólo 7). Más recientemente se ha encontrado una asociación entre el genotipo 3435TT y una menor probabilidad de fracaso virológico y aparición de resistencias en pacientes tratados con efavirenz77. Dado que el efavirenz no es sustrato de la PGP57, se plantea la posibilidad de que esta asociación se deba a un ligamiento del MDR-1 con otros genes77.
En otros estudios no se encontraron diferencias en la caída de carga viral entre los genotipos del exón 26 (C3435T) y del exón 21 (G2677T) en 31 pacientes del ensayo ACTG31586, ni se encontró relación del polimorfismo C3435T con el ascenso de la carga viral, ni el ascenso de los CD4, a los 3 y 6 meses de iniciar tratamiento antirretroviral87. Recientemente, tampoco se ha encontrado asociación entre el genotipo de los polimorfismos del exón 21 y del exón 26 y la respuesta al tratamiento76.
En cuanto a la relación entre actividad funcional de la PGP y evolución de la infección por VIH, se ha descrito en 185 pacientes con infección por VIH una relación inversa entre la actividad de la PGP y la carga viral72. En el análisis multivariable de este estudio, la actividad de la PGP no se asoció a otras variables demográficas o terapéuticas, lo que sugiere que la PGP tenía per se una actividad inhibidora sobre la replicación viral72.
Las diferencias entre los datos obtenidos in vitro e in vivo se han atribuido a que la sobreexpresión de la PGP obtenida in vitro puede llegar a ser 1.000 veces la de las células mononucleares de sangre periférica, mientras que las diferencias de la expresión de PGP entre pacientes son de dos a tres veces44.
PGP y efectos adversos de los antirretroviralesPuesto que la PGP puede influir sobre las concentraciones plasmáticas, tisulares e intracelulares de los antirretrovirales, es esperable que también pueda influir sobre la incidencia de sus efectos adversos. Partiendo de la base de que las concentraciones plasmáticas de efavirenz tienen relación con su neurotoxicidad y fracaso terapéutico88,89, se estudiaron los polimorfismos del CYP2B6 y del MDR-1 en una cohorte de pacientes VIH positivos tratados con efavirenz en la que se evaluaron de manera prospectiva las concentraciones plasmáticas y sus efectos tóxicos sobre el SNC. Se encontró una relación entre las concentraciones y la neurotoxicidad con el polimorfismo del CYP2B6, pero no con los polimorfismos del exón 21 y 26 del gen MDR-190.
Se ha comunicado recientemente que la hepatotoxicidad por nevirapina y efavirenz tiene relación con el polimorfismo C3435T (CC < CT < TT)60,91. Dado que el efavirenz no es sustrato de la PGP, la explicación de esta asociación podría ser la misma que para la asociación de este polimorfismo con la frecuencia de fracaso virológico en pacientes tratados con efavirenz, es decir, un posible ligamiento del polimorfismo del MDR-1 a otros genes77.
Se ha encontrado relación entre el polimorfismo C3435T del MDR-1 y el ascenso de las concentraciones plasmáticas de colesterol de las lipoproteínas de alta densidad (c-HDL) (menores concentraciones en genotipo TT) al año de tratamiento con efavirenz por mecanismos no bien conocidos89.
Implicaciones terapéuticasEl potencial efecto de los cambios en la expresión de la PGP sobre la farmacocinética e incluso farmacodinamia de los antirretrovirales daría pie a posibles intervenciones terapéuticas para modular su expresión y acción. La hipótesis de que un exceso de actividad de la PGP, genética o inducida, puede reducir la eficacia del tratamiento antirretroviral por disminuir el acceso de los IP a su lugar de acción, abre varias posibilidades terapéuticas.
Se podrían utilizar en los pacientes farmacorresistentes los antirretrovirales que no sean sustrato de la PGP o en el caso de utilizar antirretrovirales que sean sustrato de la PGP, asociarles otros inhibidores de la PGP para aumentar su concentración intracelular. El ritonavir puede aumentar la eficacia de otros antirretrovirales por un efecto farmacodinámico (menores resistencias primarias, efecto sinérgico con otros fármacos y menor desarrollo de resistencias secundarias), pero, sobre todo, tiene un efecto farmacocinético aumentando las concentraciones plasmáticas e intracelulares de otros IP por su efecto inhibidor del CYP3A4 y de la PGP92.
Hay diversos fármacos que actúan como inhibidores de la PGP, que se diferencian por su mayor o menor tolerabilidad y especificidad. El verapamilo y la quinidina podrían aumentar el acceso de los IP a su lugar de acción79. El biricodar93, que inhibe la PGP y la MRP-1, y el valspodar (SDZ PSC 83 3)94,95 y el zosuquidar (LY335979)96,97, que inhiben la PGP, han sido estudiados en oncología con resultados prometedores, aunque tienen también un efecto inhibidor sobre el CYP3A4 que puede dificultar su uso. El ontogen (ÜC144-093 o ONT-093) es un inhibidor de la PGP más específico, ya que no inhibe las proteínas asociadas con el gen MRP1, no es metabolizado por el CYP3A4, CYP2C8 ni CYP2C9, por lo que tiene pocas posibilidades de interacciones con los antirretrovirales, y parece bien tolerado en los ensayos oncológicos en fase I, lo que le hace un buen candidato para asociar a los antirretrovirales en los casos farmacorresistentes98-100.
La inhibición de la expresión del gen MDR-1 se plantea también como un vía para limitar las acciones de la PGP. Se han utilizado diversas estrategias, como olinucleótidos antisentido101,102, ARN de interferencia103, factores de trascripción artificiales104 y ribozimas105-107, para frenar su expresión. Sin embargo, aunque los resultados obtenidos con los inhibidores de la PGP en oncología son prometedores, en el caso de la infección por VIH se requerirían tratamientos más prolongados desconociéndose su toxicidad a largo plazo.
La utilización de todas estas vías de inhibición de la PGP puede ser un procedimiento para acceder a los santuarios donde se establece el VIH sin que pueda ser erradicado29-31, y de esta manera facilitar su control o contribuir a la tan soñada curación de esta enfermedad.

