



El carcinoma de paratiroides es una enfermedad poco frecuente que aparece en un 0,5-5% de los casos de hiperparatiroidismo primario. Se caracteriza por la combinación de intensos síntomas de hipercalcemia, muy elevadas concentraciones séricas de calcio y paratirina y masa cervical palpable. El diagnóstico de certeza se obtiene mediante el estudio histológico tras la cirugía. Presentamos a un paciente varón de 77 años de edad que ingresó en el hospital por tromboembolia pulmonar con hipercalcemia concomitante; inicialmente se etiquetó como hiperparatiroidismo primario, pero que presentaba las características clínicas atípicas descritas. Con la sospecha clínica de carcinoma de paratiroides, se realizó intervención quirúrgica y estudio anatomopatológico, que confirmó el diagnóstico de carcinoma paratiroideo.
Parathyroid carcinoma (PC) is an uncommon disease affecting 0.5-5% of all patients with primary hyperparathyroidism. PC is characterized by the association of severe symptoms of hypercalcemia, high serum calcium and parathyroid hormone (PTH) concentrations and a palpable neck mass. Definitive diagnosis can only be made by histological study after surgery. We report the case of a 77-year-old man admitted to our hospital due to pulmonary embolism and hypercalcemia. The patient was initially diagnosed with primary hyperparathyroidism, but displayed the atypical clinical features described above. Due to clinical suspicion of PC, a surgical procedure was carried out. Diagnosis of parathyroid carcinoma was confirmed by histopathologic study.
El carcinoma de paratiroides (CP) es una causa rara de hiperparatiroidismo primario, con una incidencia<1%1. Son tumores de crecimiento lento que tienden a recurrir localmente y se extienden por contigüidad a las estructuras cervicales. El tratamiento inicial de elección es quirúrgico y el estudio anatomopatológico permite realizar un diagnóstico de certeza2. Su diagnóstico diferencial con adenoma antes de la intervención quirúrgica es difícil, aunque algunos hallazgos como síntomas clínicos marcados, concentraciones muy elevadas de paratirina (PTH) y la presencia de una masa cervical palpable pueden indicar una lesión maligna3.
Se describe el caso de un paciente que ingresó en nuestro hospital por tromboembolia pulmonar (TEP) y en el que posteriormente se detectaron elevadas concentraciones séricas de calcio y PTH junto con un nódulo cervical palpable. Éste es un caso de especial interés, dada la baja frecuencia de carcinoma de paratiroides, que además se diagnosticó en un paciente de edad avanzada y de forma casual ante la presentación de tromboembolia pulmonar.
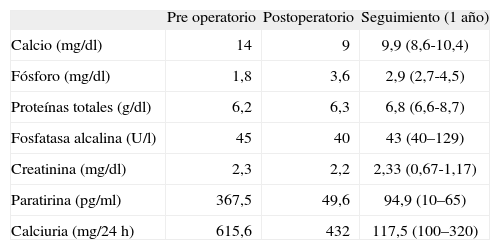
CASO CLÍNICOVarón de 77 años que consultó por un cuadro de disnea y dolor torácico pleurítico compatible con TEP secundario a una trombosis venosa profunda en la extremidad inferior izquierda. En el estudio inicial se detectó hipercalcemia, motivo por el que se consultó con nuestro servicio. Entre los antecedentes personales del paciente, destacaban un adenocarcinoma de próstata en remisión, tratado con radioterapia (RT) y hormonoterapia 2 años antes, TEP durante la RT, aneurisma de aorta abdominal intervenido quirúrgicamente (bypass aortobiilíaco) y la existencia de hipertensión arterial en tratamiento farmacológico e insuficiencia renal crónica leve. La anamnesis dirigida ante la sospecha de hiperparatiroidismo primario (HP) reveló un cuadro de un año de evolución de poliuria, polidipsia, astenia, dispepsia y estreñimiento. Asimismo el paciente había presentado un cólico nefrítico 10 meses antes del presente ingreso. En la exploración cervical destacó un nódulo de 2cm en la región infrahioidea derecha, no adherida a planos profundos. Los hallazgos analíticos iniciales se muestran en la tabla 1.
Hallazgos de laboratorio
| Pre operatorio | Postoperatorio | Seguimiento (1 año) | |
| Calcio (mg/dl) | 14 | 9 | 9,9 (8,6-10,4) |
| Fósforo (mg/dl) | 1,8 | 3,6 | 2,9 (2,7-4,5) |
| Proteínas totales (g/dl) | 6,2 | 6,3 | 6,8 (6,6-8,7) |
| Fosfatasa alcalina (U/l) | 45 | 40 | 43 (40–129) |
| Creatinina (mg/dl) | 2,3 | 2,2 | 2,33 (0,67-1,17) |
| Paratirina (pg/ml) | 367,5 | 49,6 | 94,9 (10–65) |
| Calciuria (mg/24h) | 615,6 | 432 | 117,5 (100–320) |
Se instauró tratamiento con hidratación y bisfosfonatos (pamidronato en dosis única de 0,3mg/kg) y se completó el estudio de HP. La ecografía cervical objetivó un nódulo no homogéneo y de patrón mixto de 2,6×2,7cm, con vaso central sospechoso de malignidad en relación con la glándula paratiroides inferior derecha (fig. 1A). En la ecografía abdominal no se apreciaron datos de nefrocalcinosis. La gammagrafía cervical con 99mTc-MIBI confirmó un foco de hipercaptación correspondiente a acumulación del radiotrazador en el polo inferior del lóbulo tiroideo derecho, persistente en ambas fases de la exploración y compatible con un adenoma paratiroideo (fig. 1B). La densitometría ósea lumbar fue normal para la edad y el sexo del paciente (T-score=−0,7; Z-score=−0,6).


A: ecografía cervical en la que se observa un nódulo no homogéneo y de patrón mixto de 2,6×2,7cm, con vaso central. B: gammagrafía paratiroidea con 99mTc-MIBI, en la que se observa foco de hipercaptación en el polo inferior del lóbulo tiroideo derecho, persistente en ambas fases. C: estudio macroscópico de la tumoración, lobulada, de 3cm, rodeada de seudocápsula fibrosa, con invasión vascular. D: examen microscópico, con células en patrón trabecular, con mitosis y focos de necrosis.
Tras 14 días de tratamiento médico, y con concentraciones de calcio sérico de 12,3mg/dl, fue sometido a intervención quirúrgica. Los hallazgos intraoperatorios mostraban un nódulo grande, de aproximadamente 3cm, duro, con superficie multinodular, que englobaba una estructura de aspecto glandular en su interior. Durante el acto quirúrgico se realizó exéresis del nódulo junto con el tejido tiroideo adyacente adherido por un extremo. No se apreciaron adenopatías macroscópicamente.
El estudio macroscópico de la pieza quirúrgica apreció una tumoración de 3cm, con una seudocápsula fibrosa y gran invasión vascular (fig. 1C). El estudio histológico mostró una lesión constituida por células oncocíticas con patrón trabecular, que presentaban núcleos grandes con nucléolos prominentes y frecuentes mitosis junto con algunas atipias y focos de necrosis (fig. 1D). La lesión estaba parcialmente delimitada por una seudocápsula fibrosa que en algunas áreas infiltraba la grasa periglandular. Presentaba, además, gran invasión venosa. Estos hallazgos fueron compatibles con el diagnóstico de CP.
Durante el postoperatorio el paciente presentó una rápida reducción de las concentraciones séricas de calcio y PTH, sin hipocalcemia. No precisó suplementos de calcio ni de vitamina D. En el seguimiento posterior, a los 3, 6, 9 y 12 meses, el paciente presentó normocalcemia y permaneció libre de enfermedad, sin signos clínicos, bioquímicos y radiológicos de recurrencia. En el momento actual presenta un hiperparatiroidismo secundario leve debido a insuficiencia renal crónica. Los parámetros analíticos tras 1 año de seguimiento se muestran en la tabla 1. En un estudio ecográfico cervical de control, no se objetivaron adenopatías significativas ni otras lesiones que indicaran recidiva.
DISCUSIÓNEl CP es una enfermedad poco frecuente que se presenta en un 0,5-5% de los casos de hiperparatiroidismo primario, y su incidencia en Europa es<1%1. A diferencia de los tumores benignos, no muestra predilección por el sexo femenino y aparece a una edad más temprana (en la quinta década de la vida)2.
La etiología exacta del cáncer de paratiroides es desconocida. Se han descrito múltiples factores predisponentes, aunque ninguno ha sido confirmado. Entre ellos figuran el antecedente de irradiación cervical, cuya asociación es débil, el hiperparatiroidismo familiar autosómico dominante y el síndrome HPT-JT (hereditary hyperparathyroidism-jaw tumor syndrome) que se caracteriza por la agregación de tumores paratiroideos y tumores óseos mandibulares4. Con respecto a la patogenia molecular, hay evidencia creciente de la implicación tanto de oncogenes como de genes supresores de tumor en el desarrollo de neoplasias paratiroideas. Uno de ellos es el gen del retinoblastoma en el cromosoma 13, que codifica una proteína con actividad supresora tumoral5. Diversos estudios muestran que la inactivación adquirida de este gen por deleción en un alelo contribuye al desarrollo del carcinoma. Otros genes implicados han sido el p53 (gen supresor tumoral) y el PRAD1 (cuya mutación se asocia a sobreexpresión de la proteína ciclina D1, hecho frecuente en el CP). Asimismo, se ha descrito mutaciones en el gen HRPT2 (un gen supresor de tumor localizado en el cromosoma 1) en el carcinoma de paratiroides esporádico, en el hiperparatiroidismo familiar aislado y en el síndrome HPT-JT6.
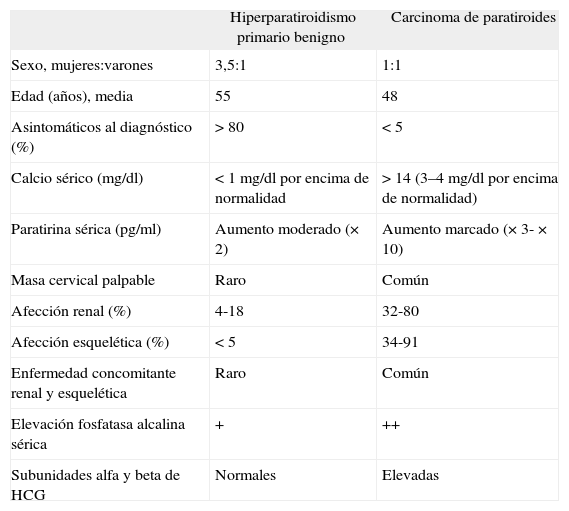
Las manifestaciones clínicas se deben principalmente a la secreción excesiva de PTH por el tumor funcionante más que a la infiltración de órganos por la masa tumoral, y por ello los signos y síntomas de hipercalcemia dominan el cuadro clínico. Pese al solapamiento de las características clínicas entre el hiperparatiroidismo primario benigno y el carcinoma de paratiroides, algunos hallazgos en su presentación podrían indicar una etiología maligna del proceso (tabla 2)3. En el caso de nuestro paciente, los datos previos a la intervención quirúrgica eran sospechosos de malignidad, ya que presentaba un cuadro marcado de hipercalcemia, una masa cervical palpable y una ecografía compatible. La existencia de una masa cervical palpable ocurre entre el 30 y el 76% de los CP y ocasionalmente se puede observar parálisis del nervio laríngeo recurrente ipsolateral.
Hiperparatiroidismo primario benigno y carcinoma de paratiroides: características
| Hiperparatiroidismo primario benigno | Carcinoma de paratiroides | |
| Sexo, mujeres:varones | 3,5:1 | 1:1 |
| Edad (años), media | 55 | 48 |
| Asintomáticos al diagnóstico (%) | > 80 | < 5 |
| Calcio sérico (mg/dl) | < 1mg/dl por encima de normalidad | > 14 (3–4mg/dl por encima de normalidad) |
| Paratirina sérica (pg/ml) | Aumento moderado (×2) | Aumento marcado (×3-×10) |
| Masa cervical palpable | Raro | Común |
| Afección renal (%) | 4-18 | 32-80 |
| Afección esquelética (%) | < 5 | 34-91 |
| Enfermedad concomitante renal y esquelética | Raro | Común |
| Elevación fosfatasa alcalina sérica | + | ++ |
| Subunidades alfa y beta de HCG | Normales | Elevadas |
HCG: gonadotropina coriónica humana.
En cuanto al estudio histológico, macroscópicamente los CP son tumores irregulares, lobulados, blancogrisáceos, de consistencia firme y duros. Son grandes,
generalmente de más de 3cm de diámetro7. En un 50% de los casos están rodeados de una cápsula blanco-grisácea, fibrosa y densa que se adhiere a los tejidos adyacentes. Aunque no hay una histopatología patognomónica de CP, Shantz et al8 establecieron unos criterios por los que se debería sospechar malignidad de una lesión paratiroidea: organización celular en capas separadas por una trabécula fibrosa densa, invasión capsular o vascular y figuras mitóticas en las células tumorales parenquimatosas.
El diagnóstico preoperatorio de carcinoma de paratiroides es extremadamente difícil y está basado principalmente en la tríada clínica, bioquímica y radiológica. El hiperparatiroidismo primario clínico junto con una masa cervical palpable y/o parálisis del nervio laríngeo recurrente indica malignidad3. El estudio citológico mediante aspiración con aguja fina no se recomienda, puesto que no proporciona un diagnóstico de certeza y la técnica aumenta el riesgo de diseminación tumoral9,10. Las técnicas de imagen (ultrasonografía, tomografía computarizada, resonancia magnética y gammagrafía) no son diagnósticas, pero permiten determinar el tamaño y la localización de las lesiones y son de utilidad para planificar una resección curativa11,12.
El tratamiento de elección del CP es quirúrgico, tanto para el tumor inicial como en el caso de recurrencias o metástasis a distancia2,13,14. Los pacientes en los que se sospecha CP antes de la intervención deben ser sometidos a exploración de las 4 glándulas y resección en bloque de la glándula paratiroidea afecta junto con el lóbulo tiroideo ipsolateral15. Técnicas más radicales no mejoran la supervivencia y se asocian con aumento del número de complicaciones postoperatorias. Si en la exploración cervical inicial se aprecia afección ganglionar, debe realizarse disección cervical radical16. Es de especial importancia evitar la rotura de la cápsula tumoral para reducir el riesgo de diseminación. En nuestro caso, en el momento de la intervención se realizó una resección de la lesión junto con la cápsula íntegra y el tejido tiroideo adyacente. En cuanto al manejo del carcinoma diagnosticado en el postoperatorio precoz tras el estudio anatomopatológico, de acuerdo con Shane, si la histopatología del carcinoma indica agresividad tumoral con invasión vascular o capsular o el paciente permanece hipercalcémico, está indicada la reexploración cervical. En ausencia de estos criterios, se acepta el seguimiento de los enfermos con determinaciones periódicas de calcio sérico y PTH3. Tras la intervención quirúrgica, la complicación más frecuente, aunque habitualmente transitoria, es la hipocalcemia. Ésta aparece en un 40- 50% de los casos, y son más susceptibles los pacientes con hiperplasia que los que presentan nódulos únicos17. En el caso descrito, el paciente no presentó hipocalcemia, posiblemente debido a varios factores: por un lado, la escasa afección ósea previa a la cirugía y, por otro, que tenía una lesión única y no una hiperplasia glandular. La rareza de este tipo de tumor no permite la realización de estudios prospectivos para examinar el efecto de la quimioterapia y/o la radioterapia, por lo que los datos al respecto se basan en los casos descritos. Aunque hay algunos casos publicados de pacientes con mejora de la supervivencia y reducción de la recurrencia tumoral con estos tratamientos, no hay evidencia suficiente para su indicación generalizada2,3.
Además del tratamiento quirúrgico, es necesario realizar un tratamiento médico dirigido al control de la hipercalcemia y sus síntomas. En la fase aguda se requiere restauración del volumen corporal con fluidoterapia intravenosa y forzar la calciuria con diuréticos de asa. En el manejo a largo plazo, los fármacos más efectivos son los bisfosfonatos, que interfieren en la reabsorción ósea mediada por osteoclastos3. También se han utilizado otros fármacos como la mitramicina, el nitrato de galio, la calcitonina, la amifostina (WR- 2721) y los calciomiméticos18.
En cuanto a la evolución natural del carcinoma de paratiroides, éste es un tumor de crecimiento lento, con escaso potencial maligno. Tiene tendencia a infiltrar estructuras adyacentes musculares, tiroides, nervio laríngeo recurrente, tráquea y esófago. Metastatiza por vía linfática y hemática, y las localizaciones más frecuentes son los ganglios cervicales (30%), los pulmones (10-40%) y el hígado (10%)2,3.
El pronóstico es variable. Las tasas de recurrencias oscilan entre el 33 y el 78% y éstas se manifiestan a los 3–5 años tras el diagnóstico inicial3. Las tasas de supervivencia son del 85% a los 5 años y el 77% a los 10 años. Su morbimortalidad se afecta principalmente por la hipercalcemia y por la magnitud de la resección tumoral inicial19.
La concomitancia de un TEP masivo en nuestro paciente no hace más que confirmar la agresividad del carcinoma de paratiroides y ampliar el espectro de presentación de esta entidad. La hipercalcemia per se induce un estado de hipercoagulabilidad según estudios realizados en animales y se han descrito casos de trastornos trombóticos en relación con hiperparatiroidismo primario agudo20. Además, la deshidratación y la hemoconcentración relacionadas con la hipercalcemia, el antecedente de radioterapia pelviana y la presencia de una neoplasia maligna condicionan estasis venosa y un estado procoagulante, mecanismos etiopatogénicos fundamentales para el desarrollo de una trombosis venosa profunda y secundariamente TEP21,22. Éste es un caso de especial interés, dada la baja frecuencia de CP, que además se diagnostica en un paciente de edad avanzada y de forma casual ante la presentación de un TEP.
El CP es, por tanto, una causa poco frecuente de hiperparatiroidismo primario. Es difícil de diferenciar de la afección benigna antes de la cirugía, dado que las manifestaciones clínicas de ambas entidades a menudo se superponen. No obstante, es de gran importancia que el CP sea considerado en el diagnóstico diferencial de la hipercalcemia dependiente de PTH, ya que la morbilidad y la mortalidad relacionadas con este diagnóstico son considerables y los resultados óptimos están relacionados con la resección tumoral completa en el momento de la intervención inicial.
