



Hyponatraemia is the most common electrolyte imbalance in neurosurgical patients. Acute hyponatraemia is particularly common in neurosurgical patients after any type of brain insult, including brain tumours and their treatment, pituitary surgery, subarachnoid haemorrhage or traumatic brain injury. Acute hyponatraemia is an emergency condition, as it leads to cerebral oedema due to passive osmotic movement of water from the hypotonic plasma to the relatively hypertonic brain which ultimately is the cause of the symptoms associated with hyponatraemia. These include decreased level of consciousness, seizures, non-cardiogenic pulmonary oedema or transtentorial brain herniation. Prompt treatment is mandatory to prevent such complications, minimize permanent brain damage and therefore permit rapid recovery after brain insult. The infusion of 3% hypertonic saline is the treatment of choice with different rates of administration based on the severity of symptoms and the rate of drop in plasma sodium concentration.
The pathophysiology of hyponatraemia in neurotrauma is multifactorial; although the syndrome of inappropriate antidiuresis (SIADH) and central adrenal insufficiency are the commonest causes encountered. Fluid restriction has historically been the classical treatment for SIADH, although it is relatively contraindicated in some neurosurgical patients such as those with subarachnoid haemorrhage. Furthermore, many cases admitted have acute onset hyponatraemia, who require hypertonic saline infusion. The recently developed vasopressin receptor 2 antagonist class of drug is a promising and effective tool but more evidence is needed in neurosurgical patients. Central adrenal insufficiency may also cause acute hyponatraemia in neurosurgical patients; this responds clinically and biochemically to hydrocortisone. The rare cerebral salt wasting syndrome is treated with large volume normal saline infusion. In this review, we summarize the current evidence based on the clinical presentation, causes and treatment of different types of hyponatraemia in neurosurgical patients.
La hiponatremia es el desequilibrio electrolítico más común en los pacientes neuroquirúrgicos. La hiponatremia aguda es especialmente frecuente en los pacientes neuroquirúrgicos después de alteraciones cerebrales de cualquier tipo, incluidos tumores cerebrales y su tratamiento, cirugía hipofisaria, hemorragia subaracnoidea o lesión cerebral traumática. Supone una urgencia, ya que origina edema cerebral debido al movimiento osmótico pasivo de agua desde el plasma hipotónico al cerebro relativamente hipertónico, que es en última instancia la causa de los síntomas asociados con la hiponatremia. Estos incluyen el descenso del nivel de conciencia, crisis convulsivas, edema pulmonar no cardiogénico o herniación cerebral transtentorial. Es imperativo el tratamiento inmediato para prevenir esas complicaciones, limitar el daño cerebral permanente y, en consecuencia, permitir una recuperación rápida después de la afectación cerebral. La infusión de solución salina hipertónica al 3%, a velocidades de administración diferentes en función de la gravedad de los síntomas y del ritmo de descenso de la concentración plasmática de sodio, es el tratamiento de elección.
La fisiopatología de la hiponatremia en los neurotraumatismos es multifactorial, aunque las causas encontradas con más frecuencia son el síndrome de antidiuresis inapropiada y la insuficiencia suprarrenal central. La restricción de líquidos ha sido tradicionalmente el tratamiento clásico del síndrome de antidiuresis inapropiada, aunque está relativamente contraindicada en algunos pacientes neuroquirúrgicos, como los que sufren hemorragia subaracnoidea. Además, muchos pacientes ingresados tienen hiponatremia de comienzo agudo y precisan infusión de solución salina hipertónica. El grupo de fármacos antagonistas del receptor de vasopresina 2, desarrollados recientemente, es una herramienta prometedora y eficaz, pero se necesitan más pruebas que lo demuestren en pacientes neuroquirúrgicos. La insuficiencia suprarrenal central, que también puede causar hiponatremia en pacientes neuroquirúrgicos, responde clínica y bioquímicamente a la hidrocortisona. El raro síndrome de pérdida de sal cerebral se trata con infusión de grandes volúmenes de solución salina normal. En esta revisión se resumen las pruebas disponibles actualmente basándose en la presentación clínica, las causas y el tratamiento de distintos tipos de hiponatremia en pacientes neuroquirúrgicos.
Hyponatraemia (plasma sodium <135mmol/l) is the most frequent electrolyte disturbance encountered in clinical practice, and it is particularly common in a variety of neurosurgical conditions. Between 15–20% of patients who are subject to traumatic brain injury (TBI) of moderate severity or worse develop hyponatraemia1,2 whereas approximately 50% of patients with non-traumatic subarachnoid hemorrhage (SAH)3,4 will develop hyponatraemia during hospitalization. Furthermore, many other neurosurgical conditions present with or develop hyponatraemia postoperatively. Around 10–20% of patients admitted with intracranial hematomas, brain tumors or pituitary surgery will develop hyponatraemia5.
Severe hyponatraemia, particularly when the onset is rapid (<48h), is a life-threatening condition, associated with increased mortality6–8. Perhaps because of the high background mortality attributable to the neurosurgical insult, a number of studies suggest that hyponatraemia is not necessarily associated with increased mortality9,10 in this setting, and indeed, hypernatraemia might be stronger predictor of mortality in neurosurgical patients11. Furthermore, there is good evidence highlighting that hyponatraemia is associated with prolonged length of hospital stay12,13. Although hyponatraemia is an important factor related to clinical outcome, its relevance is frequently overlooked in neurosurgical patients14.
In this review, we consider the differential diagnosis of neurosurgical hyponatraemia, particularly in the setting of subarachnoid hemorrhage and TBI, and offer recommendations on treatment.
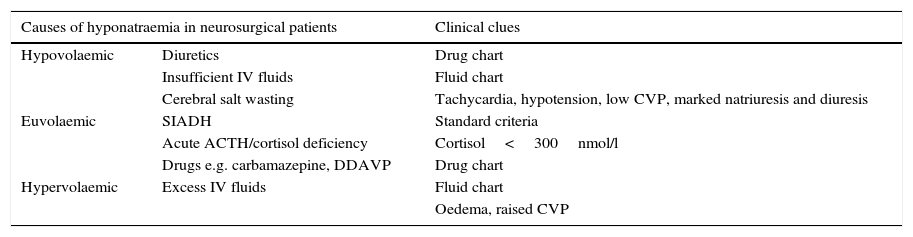
Pathophysiology and classification of hyponatraemia in neurosurgical patientsThe differential diagnosis of hyponatraemia in neurosurgical patients is simplified in Table 1. However, formulating a correct diagnosis is not always straightforward, as hyponatraemia is often multifactorial and there are multiple confounding factors which make diagnosis complex. Many patients in the neurosurgical intensive care unit are treated with large volumes of intravenous fluid, either to prevent cerebral vasospasm or to combat hypotension. The widespread use of inotropes complicate the assessment of haemodynamic factors influencing blood volume. Adrenocorticotrophin deficiency make manifest as hyponatraemia, though pituitary injury following head injury may be very transient1 and so the contribution effects on plasma sodium concentration may similarly be short-lived. As in all branches of medicine, multiple factors may contribute to hyponatraemia in a single patient. It is also important to appreciate that the cause of hyponatraemia may change in a single patient; it would not be uncommon for instance, to treat hypovolaemic hyponatraemia with appropriate intravenous fluids, only to discover underlying the syndrome of inappropriate antidiuresis (SIADH) to be simultaneously present.
- •
Hypovolemic hyponatraemia. Classical physical signs such as tachycardia, hypotension, decreased skin turgor and mucous hydration, decreased ocular pressure or osthostatism, together with the presence of raised plasma urea, are useful pointers to low effective circulatory volume due to hypovolaemia, but are not always present. If central venous pressure (CVP) is available, the diagnosis of hypovolaemia can be made more reliably. Low urinary sodium concentration is an extremely useful biochemical marker for hypovolaemia. The extremely uncommon condition of cerebral salt wasting is characterized by hypovolemia with increased sodium excretion, in the absence of other explanations for such finding (e.g. diuretic therapy or salt-wasting nephropathy).
- •
Hypervolaemia. Clinical signs of fluid overload in the appropriate context, with increased CVP, peripheral or pulmonary oedema and a positive fluid balance are the cornerstones of diagnosis of hypervolaemic hyponatraemia.
- •
Euvolaemia. SIADH is the most frequent cause of euvolaemic hyponatraemia. It is characterized by innapropriate vasopressin (AVP) release despite plasma hyposmolality, leading to water retention in the kidneys and worsening of hyponatraemia. The increase in water reabsorption is secondary to aquaporin-2 (AQP2) insertion in kidney tubules, which leads to passive water absorption through the hypertonic kidney medulla towards the systemic circulation.15 SIADH is a clinical diagnosis, supported by biochemical findings (plasma hyposmolality, inappropriate urinary concentration >100mOsm/kg, urinary sodium >30mmol/l, normal kidney and liver function) in the euvolaemic patient, and it is important to highlight it is a diagnosis of exclusion (hypothyroidism, central adrenal insufficiency). Severe hypothyroidism is an uncommon cause of euvolaemic hyponatraemia. Exclusion of ACTH deficiency is mandatory for the diagnosis of SIADH, but it is particularly important in neurosurgical patients, where cerebral trauma may damage the vascular supply of the pituitary gland. Acute ACTH deficiency has been reported in association with subarachnoid haemorrhage and intracranial haemorrhage, but most commonly following neurotrauma. The biochemical results encountered in ACTH deficiency are identical to that in SIADH, so the differentiation between the two depends on hormonal measurements. Patients with ACTH deficiency and hyponatraemia have increased plasma AVP concentrations. This is related to two different mechanisms. Firstly, hypersecretion of CRH is accompanied by the co-secretion of AVP. Secondly, hypocortisolaemia may lead to volume contraction and/or hypotension leading to baroregulated release of AVP.16 Hyponatraemia develops due to two mechanisms; the direct antidiuretic effects of increased circulating AVP, and the loss of the permissive effects of cortisol on free water excretion.17 Glucocorticoid replacement leads to normalization of plasma sodium concentration in patients with ACTH deficiency.18 The correction of hyponatraemia may be so rapid that over-correction of plasma sodium has been reported after glucocorticoid replacement.19 Acute glucocorticoid deficiency should always be considered in any patient with neurotrauma and hyponatraemia.20
Clinical clues for accurate diagnosis of hyponatraemia in neurosurgical patients.
| Causes of hyponatraemia in neurosurgical patients | Clinical clues | |
|---|---|---|
| Hypovolaemic | Diuretics | Drug chart |
| Insufficient IV fluids | Fluid chart | |
| Cerebral salt wasting | Tachycardia, hypotension, low CVP, marked natriuresis and diuresis | |
| Euvolaemic | SIADH | Standard criteria |
| Acute ACTH/cortisol deficiency | Cortisol<300nmol/l | |
| Drugs e.g. carbamazepine, DDAVP | Drug chart | |
| Hypervolaemic | Excess IV fluids | Fluid chart |
| Oedema, raised CVP | ||
IV: intravenous, CVP: central venous pressure, DDAVP: desmopressin, SIADH: syndrome of inappropriate antidiuresis.
Hyponatraemia is common following pituitary surgery, with a reported incidence of between 3 and 25%.21,22 A recent review showed that 15/129 patients dropped plasma sodium below 131mmol/l; the biggest predictor of hyponatraemia was large tumour size.23 Hyponatraemia was associated in this series with prolonged hospital stay. In most cases, SIADH is the underlying cause of post-operative hyponatraemia.5 As most patients undergoing pituitary surgery are treated with glucocorticoids to avoid perioperative ACTH deficiency, adrenal insufficiency does not usually enter the differential diagnosis. Conversely, presence of preoperative hypopituitarism is a risk factor for postoperative hyponatraemia.22 The ultimate cause of inappropriate AVP release is thought to be direct damage to the hypophyseal stalk or the posterior pituitary. Hyponatraemia is usually asymptomatic.24
Sometimes, transient hyponatraemia also occurs as part of the triple phase response,2 characterized by cranial diabetes insipidus (CDI) followed by a period of hyponatremia due to non-osmotic release of preformed AVP and finally either normalization of plasma sodium level or re-establishment of permanent CDI. Transient CDI usually occurs in the first 48h after transphenoidal resection, lasting up to 48h, whereas SIADH usually occurs 5–7 days after surgery.
The delay in onset of SIADH can lead to practical problems for neurosurgical units where there is very early discharge following pituitary surgery. A recent retrospective review of 303 patients who had undergone transsphenoidal surgery noted a 9% readmission rate, two thirds of whom were readmitted due to symptomatic hyponatraemia, a median of eight days post-surgery.25 The mean plasma sodium concentration was 119mmol/l, so this was significant hyponatraemia. As a result, some units recall patients at intervals in the week after discharge, for plasma sodium checks.
Hyponatraemia in patients following traumatic brain injuryHyponatraemia is common after traumatic brain injury, occurring in 15–20% of patients with moderate or severe injury1,5,26; 80% of cases of hyponatraemia in this setting are secondary to SIADH.2,26 Almost all cases are transient, lasting for at most the first 5 days of admission,1 and almost all resolve with improvement of the brain injury. Hyponatraemia may be longer lasting with more severe injury but chronic hyponatraemia is rare. Data from a cohort of 50 patients, who were followed prospectively for a year after traumatic brain injury (TBI) showed that 14% had hyponatraemia immediately after TBI, but none had hyponatraemia at 6 or 12 months.27
Although retrospective studies have suggested that SIADH is the principal cause of hyponatraemia following TBI, recent prospective data has raised the possibility that glucocorticoid deficiency, secondary to acute pituitary dysfunction, may be an unrecognized cause of low plasma sodium concentrations. In a large group of patients with moderate/severe TBI, who were followed prospectively with daily plasma cortisol measurements, 87% of patients with hyponatraemia had plasma cortisol concentrations of <300nmol/l (<10.8mcg/l), which were inappropriately low for the degree of hypotension/postoperative stress, and which were lower than a control group of patients who had undergone major surgery.1 All patients in this study who were treated with intravenous hydrocortisone for presumptive ACTH/adrenal insufficiency had rapid resolution of hyponatraemia.1 This data strongly suggested that transient ACTH/cortisol deficiency is much more common after TBI than had been suggested by earlier studies.28,29 Cortisol dynamics are highly variable during the acute period following TBI so multiple measurements of plasma cortisol may be necessary to identify transient deficiency.
Although the findings of this study have not been confirmed by other researchers, they would suggest that many cases of hyponatraemia following TBI which were previously thought to be due to SIADH may actually be due to acute ACTH deficiency; randomized controlled trials of the effect of steroids on outcomes in this setting would be a valuable contribution. It is also important to note that in this large prospective study, no cases of cerebral salt wasting were identified.
Hyponatraemia following subarachnoid haemorrhageHyponatraemia is very common following subarachnoid haemorrhage, occurring in approximately 50% of patients.3,4 There has been a vigorous debate in the literature as to whether hyponatraemia following SAH is due to SIADH, acute glucocorticoid deficiency or cerebral salt-wasting syndrome (CSWS). Some authors have suggested that CSWS is the commonest cause, on the basis of evidence of increased plasma levels of both brain natriuretic peptide (BNP) and atrial natriuretic peptide (ANP).30,31 However, these old studies were relatively underpowered due to small patient numbers, and not every patient who exhibited an increase in plasma BNP or ANP concentrations did in fact develop hyponatraemia. Retrospective studies failed to report CSWS as the underlying aetiology for hyponatraemia in SAH,3,5 with SIADH being the commonest cause. However, Kao and colleagues suggested from their data, taken from a retrospective cohort of SAH patients with hyponatraemia <130mmol/l, that CSWS was the underlying cause in 35% and only 22.9% of hyponatraemia was due to SIADH.32 Retrospective studies – including those from our own unit – are limited by the accuracy of data collection from neurosurgical notes and the absence of crucial diagnostic criteria for SIADH and CSWS. In addition, there has been little reporting of cortisol dynamics, so there is little appreciation of the potential contribution of acute glucocorticoid insufficiency, although it has been reported up to 12% immediately following SAH in one paper.33
In a prospective study conducted in our own centre, repeated sequential measurements of sodium and plasmatic cortisol concentrations, showed that 49% of patients with SAH developed hyponatraemia, and in 71% of cases this was due to SIADH.4 In 8.2% of hyponatraemic patients, ACTH insufficiency was revealed, and in these patients, parenteral hydrocortisone led to normalization of plasma sodium concentration. We found no data to support a single diagnosis of CSWS. Although plasma concentrations of BNP rose following SAH, they did so in nearly all patients and plasma BNP levels were not different between hyponatraemic and eunatraemic patients. In those with hyponatraemia, BNP levels were not different during or after the episode of hyponatraemia. Thus, the rise in plasma BNP concentrations was unrelated to either the development or the resolution of hyponatraemia. In contrast, plasma AVP concentrations were significantly higher in those with clinical diagnosis of SIADH compared to those who had hypovolaemic hyponatraemia, ACTH insufficiency and those with excessive fluid administration (Fig. 1).

Comparison of AVP levels between different patient groups in patients with hyponatraemia following SAH: each point represents an individual AVP measurement; adapted from4 with permission. *p=0.01; *** p<0.0001; SIAD: hyponatraemia due to SIADH; Cort Def: hyponatraemia due to acute glucocorticoid insufficiency; Hypovol: hypovolemic hyponatraemia; Fluids: hyponatraemia due to inappropriate hypotonic intravenous fluid administration; NormoNa: normal plasma sodium levels following SAH.
In summary, although BNP was elevated in almost all patients with SAH, AVP was elevated in those with hyponatraemia, and plasma levels were higher in those with clinical diagnosis of SIADH. The biochemical and clinical correlation drawn from these data suggests that SIADH is the commonest cause of hyponatraemia following SAH. Despite robust methodology and sequential measurements of both BNP and AVP, no episodes of CSWS were encountered in the largest prospective study to date. Equally, none of the patients developed permanent hyponatraemia following SAH, which suggests that hyponatraemia is a transient phenomenon.
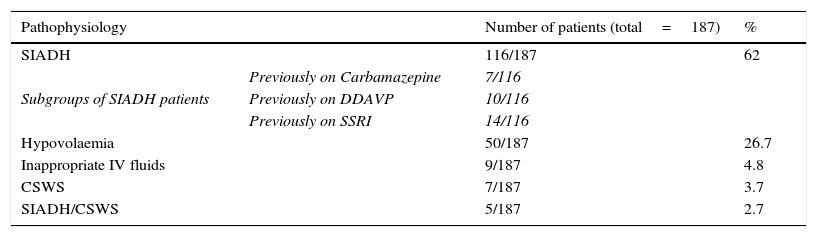
Does CSWS exist?CSWS is an uncommon cause of hyponatraemia in neurosurgical patients, which, in its classical manifestation, is characterized by hypovolaemic hyponatraemia secondary to marked diuresis and natriuresis. CSWS was first described by Peters et al. on the basis of three patients with hypovolaemic hyponatraemia,34 evidence of increased diuresis and natriuresis and normal hypothalamic, pituitary and adrenal function. CSWS was later associated with certain conditions such as TBI, SAH and clipping of intracranial aneurysms.26,35,36 Initial evidence supporting this syndrome was based on retrospective data with Nelson et al. describing 10 patients with evidence of hyponatraemia, increased natriuresis, inappropriate urine concentration and hypovolaemia.35 Two subsequent studies provided evidence for a syndrome which was distinct from SIADH.37,38 Our own data would support the view, expressed by others,39 that cerebral salt wasting is a relatively uncommon cause of neurosurgical hyponatraemia. In a retrospective survey of 1698 patients admitted to our neurosurgical unit over a twenty month period, we found that SIADH was responsible for 62% of cases of hyponatraemia (plasma sodium <130mmol/l), as outlined in Table 2.5 Cerebral salt wasting was estimated to occur in only 5% of cases, but in a retrospective case note analysis, data collection was incomplete. Although our subsequent prospective studies were unable to identify any cases of CSWS, we do not agree, however, that cerebral salt wasting simply represents an escape from antidiuresis following SIADH. The dramatic diuresis and natriuresis in cerebral salt wasting is very different from the modest responses observed in escape from antidiuresis in SIADH secondary to pulmonary disease, for example. However, the incidence of true CSWS is very rare.
Aetiology of 187 cases of hyponatraemia (plasma sodium<130mmol/l) documented in 1698 admissions to Beaumont Hospital neurosurgical unit between January 2002 and September 2003.5
| Pathophysiology | Number of patients (total=187) | % | |
|---|---|---|---|
| SIADH | 116/187 | 62 | |
| Subgroups of SIADH patients | Previously on Carbamazepine | 7/116 | |
| Previously on DDAVP | 10/116 | ||
| Previously on SSRI | 14/116 | ||
| Hypovolaemia | 50/187 | 26.7 | |
| Inappropriate IV fluids | 9/187 | 4.8 | |
| CSWS | 7/187 | 3.7 | |
| SIADH/CSWS | 5/187 | 2.7 | |
SIADH: syndrome of inappropriate antidiuresis, DDAVP: desmopressin, SSRI: selective serotonin reuptake inhibitor, IV: intravenous, CSWS: cerebral salt-wasting syndrome.
The key issues in establishing whether treatment for hyponatraemia is necessary are
- 1.
What is the cause of the hyponatraemia? Hypovolaemic hyponatraemia almost always needs treatment in SAH, as cerebral vasospasm leading to cerebral ischaemia, is an extremely adverse situation. In addition, it is difficult to withhold steroid replacement if there is clear evidence of ACTH/cortisol deficiency. On the other hand, most cases of SIADH are both mild and transient, and in the absence of good trial data, it is debatable how important active management of hyponatraemia due to SIADH is in this situation. Some authors have specifically argued against treatment.40
- 2.
How rapid is the onset of hyponatraemia? Acute hyponatraemia (<48h duration) is far more likely to cause symptoms of cerebral irritation, as cerebral oedema is more likely to occur. Almost all neurosurgical hyponatraemia is of acute onset, and the rapidity of drop in plasma sodium, along with collateral cerebral irritants such as space-occupying lesions, ischaemia/hypoxia, surgical intervention and infection, means that there is a significant likelihood that neurological symptoms requiring active intervention will develop.
- 3.
Are there symptoms of cerebral irritation? The common agreement in recently published guidelines19,41 is that emergency treatment of hyponatraemia should be driven by the presence of symptoms and signs of cerebral irritation, rather than by the severity of hyponatraemia. In clinical practice, the presence of other co-irritants as outlined in the previous paragraph dictates that neurosurgical patients may need active intervention at relatively moderate levels of hyponatraemia. Equally, in some neurosurgical patients the exact impact of hyponatraemia to the patient's status is difficult to ascribe due to presence of other concomitant factors (i.e. sedation). When the clinical condition cannot be accurately evaluated, we recommend correcting acute hyponatraemia with 3% hypertonic saline, or if the level of plasma sodium is lower than 125mmol/l. The underestimation of the effect of hyponatraemia in this setting exposes patients to unacceptable risks.
Acute hyponatraemia leads to an osmotic water shift from the hypotonic plasma to the comparatively hypertonic brain, producing cerebral oedema. The clinical manifestations are sometimes masked in patients with neurosurgical conditions, and vary from with nausea, general malaise and headaches, to seizures, reduced level of consciousness, coma and finally respiratory arrest. The risk of cerebral oedema increases with the rapidity in drop in plasma sodium concentration, but there is no threshold for one specific symptom and the progression can be rapid and unpredictable. Fatal transtentorial herniation is the main concern in acute hyponatraemia, with a higher risk in patients with intracranial pathology.42 Noncardiogenic pulmonary oedema also complicates acute hyponatraemia and its presence should alarm the physician as it is a prelude to cardiac arrest.43 Acute hyponatraemia is a medical emergency requiring immediate therapy to minimize the risk for neurological disability and mortality.
The aim of treatment in patients with acute severe hyponatraemia is the rapid elevation of plasma sodium level by 4–6mmol/l over 4h; this leads to a reduction in intracranial pressure and resolution of symptoms of herniation in nearly 50% in an hour.44 In practice, this can be achieved by the administration of a 100-ml bolus of 3% saline over 15min, repeated to a maximum of 3 times if no improvement is evident; ideally this should be supervised by a physician who is experienced in the treatment of hyponatraemia. Recent expert panel recommendations from the US19 and from Spain,45 agree on the use of 100-ml as the infused volume. There is evidence of successful treatment with this approach in a small cohort of runners with acute symptomatic exercise-induced hyponatraemia,46 though to date there are no published data relating to the use of this treatment in neurosurgical hyponatraemia.
For patients with mild or moderate symptoms, 3% saline infusion at a rate of 0.5–2ml/kg/h is the initial approach9,45 with frequent measurements of plasma sodium. The recommended target of correction is <8mmol/l in any 24h period if duration of hyponatraemia is chronic or is unknown, but the accepted maximum rise is 12mmol/l in 24h in the absence of other major risk factors for osmotic demyelination syndrome (ODS). For those at very high risk of ODS and chronic hyponatraemia or unknown duration, the recommended target is <6mmol/l with maximum accepted of 8mmol/l rise in 24h.19
If ODS is documented or suspected, decreasing plasma sodium level is the next step as this has been shown in animal models and isolated human clinical reports to reduce permanent brain damage and disability.47,48 The use of intravenous dextrose, parenteral desmopressin or both together simultaneously can be very helpful in reducing plasma sodium concentration. Our approach for reversal of overcorrection is similar to those suggested by the American Guidelines; considering 2mcg of intravenous desmopressin with concomitant use of 5% intravenous dextrose at 3ml/kg/h with hourly plasma sodium being checked until the goal is achieved.19
Management of ACTH deficiency in patients with hyponatraemiaCorrect diagnosis of ACTH deficiency is difficult in view of the lack of a gold standard test for central adrenal insufficiency (AI) in ill patients. In the presence of normal binding proteins in plasma, a cutoff of 15mcg/dl (414nmol/L) has been proposed for the diagnosis of ACTH deficiency in intensive care unit (ICU) patients.49 Conversely, other societies such as the Critical Care Medicine Taskforce suggested a random total cortisol <10 mcg/dl (284nmol/l) or a serum cortisol <9mcg/dl (250nmol/L) after ACTH administration (250mcg) for an accurate diagnosis.50 As these recommendations are for ICU patients, they seem applicable to acutely ill patients with neurosurgical conditions. Our clinical practice for patients with euvolaemic hyponatraemia and biochemical evidence of SIADH is for routine measurement of 09:00h plasma cortisol levels. We would empirically commence parenteral hydrocortisone while waiting for laboratory results in patients with clinical suggestions of cortisol deficiency, such as hypoglycemia or hypotension. In those with 9 am plasma cortisol >16.2mcg/dl (450nmol/l) ACTH deficiency can be excluded with high probability, and usually this means that SIADH is the underlying cause of hyponatraemia. We recommend hydrocortisone replacement when 09:00h plasma cortisol is <10.8mcg/dl (300nmol/l): our own published data show that levels below this cut off are inappropriately low for the degree of illness,1,4 and this cut off is in close agreement to that recommended by the Critical Care Medicine Task Force.50 In patients with intermediate plasma cortisol concentrations of between 10.8–16.2mcg/dl (300–450nmol/l), a decision should be made based on the presence of signs or symptoms of glucocorticoid deficiency (i.e. hypotension, hypoglycemia, nausea). Given the fact that the circadian rhythm of cortisol secretion is usually lost in severely stressed patients, it is probably unnecessary to standardize a 09:00 time for measurement of plasma cortisol, but our ongoing research protocols dictate that this is the time that we select.
If euvolaemic hyponatraemia is secondary to ACTH deficiency; treatment with hydrocortisone leads to rapid recovery of plasma sodium, usually within 24h, and significant improvements in other signs of glucocorticoid insufficiency are encountered. Prospective studies in our unit have shown that ACTH deficiency is frequently transient in patients with TBI1,51 and almost always following SAH2,52,53; highlighting the need for revaluation when brain insult recovers. Only those with clear evidence of ACTH deficiency at 3–6 months following a dynamic evaluation of hypothalamic-pituitary adrenal axis function are continued on long term hydrocortisone replacement.
Non-emergency treatment of hyponatraemiaAlthough most neurosurgical patients develop hyponatraemia acutely, some patients present with chronic hyponatraemia and others are less symptomatic; in these patients, alternatives other than 3% hypertonic saline can be considered.
Fluid restrictionFluid restriction is recommended as first line treatment for hospitalized patients with SIADH in both the US recommendations and European guidelines.19,41 As the evidence base for water restriction is so sparse, it is no surprise to learn that the evidence base in neurosurgical conditions is non-existent. Indeed, neurosurgeons are reluctant to sanction the use of fluid restriction, particularly in patients with SAH, because of the perceived risk of cerebral vasospasm due to hypotension. In fact, patients are routinely administered intravenous fluids to prevent blood volume contraction, at volumes which negate any attempt at fluid restriction.54 Because of neurosurgical reluctance to consider fluid restriction, pharmacological interventions are more often considered in neurosurgical patients.
UreaTreatment with oral urea has been proposed as another practical option for SIADH. Urea has been proven to be effective in SIADH in different clinical settings.55 An uncontrolled retrospective review of the treatment of SIADH in patients after SAH showed that urea normalized plasma sodium at low cost and with few documented side effects.56 In this study, plasma sodium took a median of 3 days to normalize; as the median duration of hyponatraemia following SAH is 3 days,4 the lack of a control group means it is difficult to ascertain whether the correction of hyponatraemia was secondary to the urea or if it simply represents the natural history of transient SIADH after SAH. Urea remains unlicensed for the treatment of hyponatraemia and is not available as a preparation for treatment, for these reasons, and the lack of prospective, controlled, randomized studies, we have little experience with this therapy.
Vasopressin receptor antagonists (vaptans)Vasopresin-2 receptor (V2R) antagonists represent a useful and potent tool to target hyponatraemia due to SIADH, though there is no prospective randomized data in neurosurgical patients. Vaptans act by competing for V2R binding leading to increased aquaresis and reduced urine osmolality, without effecting electrolytes metabolism. In Europe, only oral tolvaptan has been approved to be used in SIADH. There is good evidence in terms of efficacy with tolvaptan in SIADH from randomized, prospective and controlled trials over placebo.57,58 However, data supporting the use of tolvaptan for SIADH in neurosurgical patients is scarce. We have used oral tolvaptan in a patient with SAH complicated by symptomatic SIADH, and reduced Glasgow Coma Score (9/15); steady improvement in plasma sodium concentration over 48h was associated with improved cognition, such that the patient was able to engage with rehabilitation and be discharged from hospital.59 Nevertheless, there is need for new prospective studies with oral tolvaptan compared to other established therapies for SIADH in the neurosurgical setting.
Other treatmentsDemeclocycline and oral frusemide plus salt supplementation have both been used in clinical practice to treat SIADH, but neither enjoys the support of a good evidence base.
In summary, hyponatraemia is a frequent complication in neurosurgical patients. In view of the potential consequences of incorrect or delayed treatment, accurate diagnosis and specific treatment of the cause of hyponatraemia by the attending physician is mandatory to obtain optimal clinical outcome.
Conflict of interestThe authors declare no conflict of interest.
