








INTRODUCCIÓN
La mitocondria es una unidad funcional encontrada en el citoplasma de todas las células eucariotípicas del organismo. La mitocondria efectúa la mayor parte de las oxidaciones celulares y produce el conjunto del adenosintrifosfato (ATP) de las células animadas en 3 etapas: oxidación mitocondrial, el ciclo de Krebs y la cadena respiratoria1. Los defectos mitocondriales intervienen en diversas enfermedades metabólicas y neurodegenerativas, así como en el envejecimiento. El denominador común de las citopatías mitocondriales es la existencia de un desorden de la cadena respiratoria que puede estar directamente en relación con una mutación del ADN mitocondrial. Ésta puede ser primitiva o consecutiva a una mutación de un gen nuclear, o secundaria a un déficit de fosforilación oxidativa inducida por el genoma del núcleo celular2,3. En este último caso existen enfermedades degenerativas con un papel preponderante de la apoptosis como en la ataxia de Friederich3,4. Alteraciones mitocondriales han sido igualmente señaladas por Blanche et al5 y Poultron et al6 en niños de madres seropositivas para el virus de la inmunodeficiencia humana (VIH) tratadas con análogos nucleósidos, especialmente la zidovudina (AZT) y la lamivudina (3TC). Hay que señalar que las series americanas7 no han confirmado estas observaciones, y es posible que los análogos nucleósidos sean tóxicos para las mitocondrias de los adultos y de los niños, inhibiendo la gammapolimerasa, la enzima que duplica el ADN mitocondrial.
Los rasgos clínicos de las enfermedades provocadas por los defectos genéticos de las mitocondrias se reflejan a menudo en los tejidos que precisan gran cantidad de energía, como los sistemas nerviosos central y periférico, el ojo, el músculo, el riñón y los órganos endocrinos. La edad y la forma de aparición y el curso clínico de las enfermedades mitocondriales varían enormemente debido al mecanismo tan poco habitual de replicación del ADN mitocondrial (ADNmt), que difiere del correspondiente al genoma nuclear. Muchas enfermedades mitocondriales se caracterizan por un modo de herencia materna, ya que el ADNmt se transmite con el ovocito. Desde que se descubrió la primera mutación del ADNmt en 1988 se han descrito cientos de mutaciones distintas8,9.
ESTRUCTURAS Y FUNCIÓN DEL ADN MITOCONDRIAL
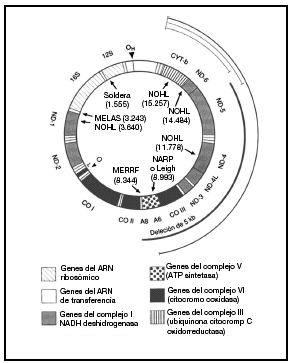
Casi todas las células contienen varios cientos de mitocondrias, aunque su número varía según las necesidades energéticas y la función de los tejidos. Las mitocondrias son los únicos orgánulos celulares que poseen su propio ADN extracromosómico. El ADNmt humano es una pequeña (16.569 pb) molécula redonda de doble cadena que codifica 16 subunidades proteicas de 4 complejos bioquímicos distintos de fosforilación oxidativa. El ADNmt codifica además los 24 ARN estructurales (2 ARN ribosómicos y 22 ARN de transferencia) necesarios para la traducción mitocondrial de estas proteínas. El bucle D (desplazamiento) no codificador es una región reguladora que controla la transcripción y la replicación. Se conocen mutaciones del ADNmt de todos los tipos de genes mitocondriales (fig. 1)10.
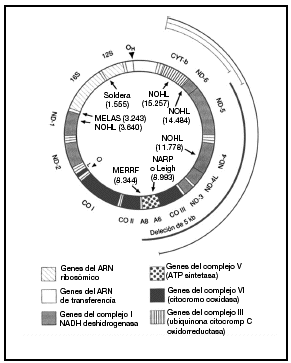
Fig. 1. Diagrama del ADN mitocondrial humano y de las mutaciones patogénicas más destacadas. Las mutaciones puntuales de los genes codificados de proteínas y de los agentes estructurales se presentan en la parte interna del círculo junto al fenotipo clínico y el nucleótido donde reside la mutación. El arco grueso indica la posición de la deleción única más común, que mide 5 kb, y los arcos finos en el interior del círculo representan las deleciones múltiples. MERRF: epilepsia mioclónica y episodios afines al ictus; NOHL: neuropatía óptica hereditaria de Leber; NARP: neuropatía, ataxia y retinitis pigmentosa; Leigh: enfermedad de Leigh heredada de la madre. Tomada de Johns10.
Resulta probable que las mitocondrias evolucionaran a partir de microorganismos independientes que se incorporaron a la célula mediante endosimbiosis. Como resultado, las mitocondrias replican, transcriben y traducen su ADN con independencia del ADN nuclear, aunque las funciones de la célula y de la mitocondria son interdependientes. Las proteínas codificadas por el ADN nuclear también intervienen en la fosforilación oxidativa y los innumerables compuestos macromoleculares necesarios para la estructura y la función mitocondriales (p. ej. replicación, transcripción y traducción del ADNmt) se importan del citoplasma a las mitocondrias9.
Las principales funciones de las mitocondrias son la fosforilación oxidativa y la síntesis del ATP para los procesos celulares que necesitan energía. Las alteraciones de la función mitocondrial provocan enfermedades mediante 3 mecanismos:
1. Reducción del aporte de ATP cuando las mutaciones afectan a la fosforilación oxidativa.
2. Generación de moléculas de oxígeno reactivo, como H2O y radicales de OH libre que pueden dañar el ADN, las proteínas o los lípidos.
3. Ejecución de la apoptosis cuando las mitocondrias liberan factores que favorecen la muerte celular, como caspasas, citocromo C y factor inductor de la apoptosis.
El ADNmt posee ciertas características clínicas únicas que lo hacen vulnerable a las mutaciones y contribuyen a la enfermedad. Por ejemplo, el ADNmt carece de intrones (por consiguiente, una mutación aleatoria afecta a menudo a una secuencia codificadora del ADN) y de histonas protectoras; posee, además, un sistema imperfecto de reparación del ADN y se encuentra expuesto a los radicales de oxígeno libre generados por la fosforilación oxidativa. Se calcula que el ADNmt se hereda exclusivamente de la madre y no se recombina y que las mutaciones del ADNmt se acumulan de forma secuencial a lo largo del linaje materno. Estas propiedades han convertido las variaciones de las secuencias de ADNmt en una herramienta de valor incalculable para los biólogos expertos en evolución y los médicos forenses.
Cada mitocondria contiene entre 2 y 10 moléculas de ADNmt y cada célula posee diversas mitocondrias (poliplasmia). La genética mitocondrial se rige por los principios de la genética de población y no de la mendeliana. Cuando surge una nueva mutación del ADNmt, las células albergan en un primer momento copias de secuencias de ADN normal y mutante estado denominado heteroplasmia que permiten que una mutación, por lo demás letal, persista y cause enfermedad. La presencia de ADNmt completamente normal o completamente mutante recibe el nombre de homoplasmia. Durante la división celular las mitocondrias se reparten de forma desigual entre las células hijas mediante el proceso denominado segregación replicativa, con lo que el porcentaje de moléculas de ADNmt mutante y normal varía. La presión de la selección opera en los niveles molecular, celular y orgánico. El porcentaje de ADNmt mutante necesario para expresar un fenotipo nocivo se denomina efecto liminar; varía de una persona a otra, entre los distintos órganos y dentro de un mismo tejido, en función de un delicado equilibrio entre la oferta y la demanda oxidativa. Estos rasgos de la segregación del ADNmt, unidos a la transmisión desigual de las mitocondrias de las células hijas durante la división celular, sientan las bases de la diversidad fenotípica apreciadas en las enfermedades mitocondriales11.
MITOCONDRIOPATÍAS
El diagnóstico de mitocondriopatía ha sido evocado ante las alteraciones neuromusculares iniciadas de forma más o menos inexplicada, después de signos derivados de la disfunción de órganos aparentemente sin relación mutua que finaliza en un cuadro clínico rápidamente progresivo.
La afección puede alcanzar a órganos múltiples, como los músculos esqueléticos, el sistema nervioso central, el ojo (atrofia óptica, oftalmoplejía externa progresiva), el oído (sordera), el sistema digestivo (insuficiencia hepática, atrofia vellositaria intestinal), el riñón (insuficiencia renal por tubulopatía proximal de Toni-Debré-Fanconi), el corazón (cardiomiopatía) y el sistema endocrino (diabetes)12-14.
Raros casos de asociación de una citopatía mitocondrial materna concomitante con un embarazo han sido descritos. Se trata de déficit en carnitina palmitoiltransferasa15 y de alteraciones de la cadena respiratoria como la oftalmoplejía externa progresiva16,17, el síndrome MELAS18-20 y el déficit en citocromo-oxidasa21.
OBSERVACIÓN
Presentamos el caso de una paciente de 30 años de edad, de origen magrebí, G3P0, ingresada en la Unidad de Alto Riesgo Perinatal (Gestaciones Patológicas), con 26 semanas de amenorrea. Entre sus antecedentes medicoquirúrgicos hay que señalar un asma que no precisa tratamiento médico, un carcinoma papilar de tiroides que fue tratado con tiroidectomía total, yodo radiactivo y tratamiento sustitutivo con hormonas tiroideas, una embolia pulmonar y una mitocondriopatía diagnosticada a los 16 años de edad. El diagnóstico se realizó ante la aparición de una insuficiencia renal por nefropatía tubulointersticial, después de neuropatía óptica bilateral y de una leucoencefalopatía, con numerosos hipersignos diseminados de manera bilateral por resonancia magnética nuclear. La biopsia muscular con su resultado histológico, inmunohistoquímico, inmunoenzimático y microelectrónico (visualización de mitocondrias anormales) confirmó el diagnóstico. El análisis espectrofotométrico, en efecto, descubrió un déficit combinado de los complejos I, II, III y IV, sin aumento de la citratosintetasa. Por el contrario, las investigaciones genéticas no han encontrado ninguna de las mutaciones ya descritas, no invalidando por otra parte el diagnóstico efectuado por las pruebas histológicas e histoquímicas irrefutables.
Los antecedentes ginecológicos reflejan una primera gestación extrauterina izquierda tratada por salpingectomía y un aborto espontáneo.
El primer trimestre de la actual gestación se desarrolla sin incidente. La ecografía de 12 semanas de amenorrea encuentra una translucencia nucal de 1 mm para una longitud craneocaudal de 54 mm. El examen bioquímico es considerado patológico en razón de una fracción de gonadotropina coriónica humana (HCG) de 9,93 µmol, pero el cariotipo fue normal en el estudio de los amniocitos. No se realizó ningún estudio enzimático fetal en ese momento de la gestación. Una ecografía realizada en la semana 22 de amenorrea no mostró ninguna anomalía morfológica fetal; los parámetros biométricos fueron satisfactorios. La velocimetría Doppler mostraba un índice de resistencia umbilical normal (IR = 0,69), pero la de las arterias uterinas eran patológicas con señales protodiastólicas bilaterales y resistencias elevadas (IR = 0,67 y 0,64). La paciente tenía una tensión arterial mantenida con Tenormin® y después Labetalol, 2 comprimidos por día. Se le suministró anticoagulación en dosis preventiva (Clexane®, 40 mg diarios por vía subcutánea), por sus antecedentes de embolia pulmonar. Progresivamente surgieron las complicaciones:
La hipertensión arterial se iba agravando gradualmente, lo que obligó a realizar un tratamiento más intensivo: Tenormin® 100 mg/día, después biterapia con Tenormin® 100 mg/día y Labetalol 400 mg/día, después Catapresan® 0,30 mg/día y Labetalol 400 mg/día.
Por el contrario, la afección ocular permanecía estable. El examen oftalmológico, realizado en el curso del embarazo, no mostró ninguna anomalía sobreañadida.
La afección neurológica central permaneció asintomática.
En la semana 25 de amenorrea, se detecta una diabetes gestacional, que rápidamente se transforma en dependiente de insulina (Actrapid® 10 U, 6 U, 5 U por día), complicada por una crisis hiperglucémica severa en el curso de su hospitalización.
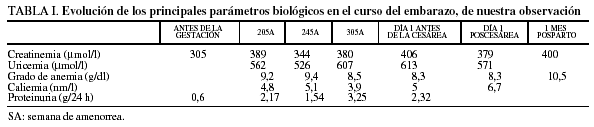
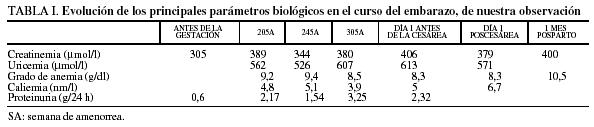
La insuficiencia renal se agrava regularmente y se complica por una preeclampsia subaguda (tabla I).
Hemos podido contar con las cifras de lactacidemia dosificada en la semana 20 de amenorrea: ácido láctico en reposo de 1,7 mmol/l; 1 h después del desayuno era de 3,2 mmol/l y 20 min después de la prueba de esfuerzo era de 6,9 mmol/l (los valores normales oscilan entre 0,8 y 1,2 mmol/l).
Sobre el lado fetal, se considera que la vitalidad es satisfactoria, especialmente reflejada en los registros de ritmo cardíaco fetal.
Después de su admisión en el servicio, a la semana 26 de amenorrea, se somete a la paciente a corticoterapia (Celestone, 12 mg, 2 días consecutivos). Las ecografías de vigilancia o seguimiento reflejan biometrías en el décimo percentil, Doppler uterinos patológicos (IR > 0,60 con señales protodiastólicas bilaterales), en tanto que los Doppler fetoplacentarios permanecían estables. Teniendo en cuenta el progreso del embarazo y las incertidumbres del diagnóstico prenatal por los métodos enzimáticos, la paciente no ha querido someterse a una nueva amniocentesis.
En la semana 30 de amenorrea, la paciente comienza con cefaleas y vómitos, la tensión arterial alcanza 200/120 mmHg, rebelde a todo tipo de terapia, con oliguria e hipercaliemia de 6,7 mmol/l, a pesar del tratamiento con Kayexalato® 60 g/día. Se realizó la extracción fetal de urgencia por cesárea con raquianestesia en la semana 30 de amenorrea + 2 días; nació una niña de 1.070 g, con test de Apgar de 1 al min y 5 a los 5 min de vida (pH arterial, 7,39; pH venoso, 7,44).
La evolución de la niña inmediatamente al nacimiento y a las semanas siguientes fue satisfactoria.
La paciente fue remitida a la Unidad de Vigilancia Intensiva (UVI), donde la tensión arterial se equilibró progresivamente con tratamiento médico, así como la diuresis, con estricta vigilancia del manejo de diuréticos. La función renal a los 7 días de la intervención era de 520 µmol/l de creatinina, 27,4 nmol/l de urea y una caliemia de 4,3 mmol/l. La vigilancia constante de la glucemia ha permitido suspender la insulina a partir del sexto día del posparto.
Después de esta gestación, la situación nefropática permanecía relativamente precaria, lo que obligó a la paciente a someterse a sesiones de diálisis, al ritmo de 3 veces por semana. Se la incluyó en el programa de trasplante renal. En el postoperatorio un nuevo elemento clínico se sobreañadió, puesto que aparecie-ron crisis convulsivas, que fueron tratadas con Neurontin®. En el plano terapéutico específico de la mitocondriopatía, la paciente sigue un tratamiento con Levocarnitina®.
COMENTARIOS Y DISCUSIÓN
Mutaciones del ADN mitocondrial
Las mutaciones del ADNmt que originan alteraciones graves y letales de la fosforilización oxidativa como defectos estructurales macroscópicos o mutaciones puntuales del ADN estructural sólo resultan viables con carácter heteroplásmico. En cambio, la mayoría de las mutaciones del ADNmt, más leves y de sentido erróneo, que afectan a los genes codificadores de proteínas son homoplásmicas. Se han descrito mutaciones puntuales del ADNmt para todos los tipos de genes mitocondriales, pero en los fenotipos de la encefalomiopatía mitocondrial mutisistémica predominan las mutaciones del ARNt; en cambio en la neuropatía óptica hereditaria de Leber (NOHL) destacan las mutaciones de los genes codificadores de proteínas. Una mutación puntual en el gen del ARN ribosómico 12S se asocia con la sordera sensitivonerviosa tanto espontánea como asociada a los aminoglucósidos (fig. 1)10.
Resulta complicado establecer una relación definitiva de causa-efecto entre una mutación del ADNmt y un fenotipo clínico por las siguientes razones9:
El ADNmt es sumamente polimorfo.
Distintas mutaciones pueden asociarse con el mismo fenotipo o la misma mutación puede asociarse con diversos fenotipos.
Los factores epigenéticos repercuten en las manifestaciones clínicas.
Enfermedades mitocondriales
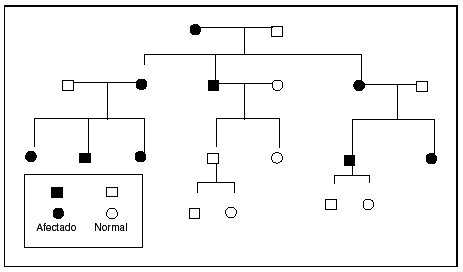
El fenómeno de la herencia materna se puede identificar cuando la mutación causante reside en el genoma mitocondrial. El pequeño genoma mitocondrial contiene 13 polipéptidos que representan subunidades de enzimas implicadas en la fosforilación oxidativa. Este genoma se hereda por completo por vía materna; las mutaciones del ADNmt se transmiten de una madre afectada a todos sus hijos con independencia del sexo22,23. En la segregación durante la mitosis, las mitocondrias se comportan como poblaciones, de manera que si la mutación no se encuentra presente en la totalidad de los genomas mitocondriales, tampoco se encontrará presente en algunas células de la progenie. Esta segregación explica la variablidiad clínica asociada con estas condiciones. Los principales trastornos mitocondriales heredados por vía materna son los siguientes: la neuropatía óptica de Leber, un trastorno caracterizado por pérdida de la visión central y arritmias cardíacas asociado como mínimo con una mutación específica del ADNmt24. La encefalopatía mitocondrial, la acidosis láctica y los síntomas similares a accidente cerebrovascular (MELAS) y la epilepsia mioclónica con fibras rojas rasgadas (MERRF) se asocian con mutaciones heredadas por vía materna25-27. El diagnóstico se establece mediante el examen microscópico y estudios moleculares del tejido muscular en los pacientes afectados. El patrón de herencia se ilustra en la figura 223.
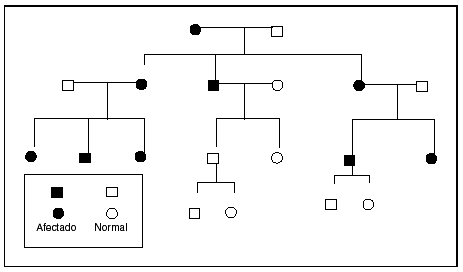
Fig. 2. Prototipo del árbol genealógico de herencia mitocondrial. Obsérvese que la alteración puede afectar tanto a los varones como a las mujeres. El trastorno afectará a toda la progenie de una madre afectada y no a la progenie de los varones afectados. Tomada de Seashore23.
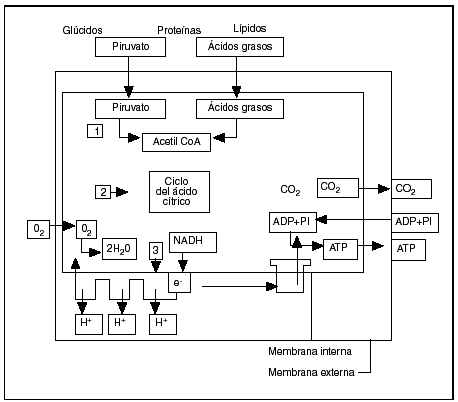
Se reagrupan, con el nombre de enfermedades mitocondriales, diferentes alteraciones orgánicas cuyo punto común radica en la disfunción de la fosforilación oxidativa. La mitocondria es, en efecto, el órgano de producción de energía de todas las células eucarióticas (fig. 3).
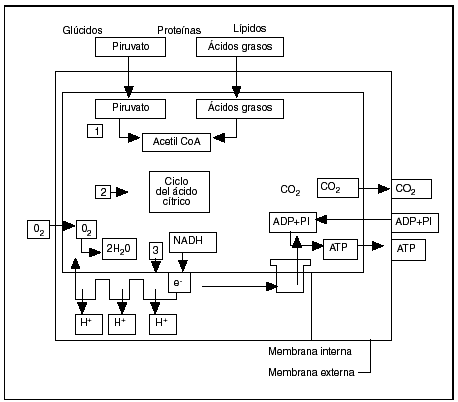
Fig. 3. Esquema de la fosforilación oxidativa en la mitocondria (según Alberts et al1). Primera etapa: la producción de acetilcoenzima A. Segunda etapa: el ciclo de Krebs. Tercera etapa: la cadena respiratoria.
La primera etapa que se produce en la matriz mitocondrial es la oxidación que acaba en la producción de acetilcoenzima A por transformación de los nutrientes inicialmente presentes en el citoplasma de la célula, bien a partir del ácido pirúvico proveniente de la glucólisis o de la degradación de los ácidos aminos por la vía de la desaminación o de la transaminación, o bien a partir de los ácidos grasos según las reacciones en hélice de Lynen. Es preciso señalar que, para atravesar la membrana interna del ácido graso, se ha debido acoplar a la carnitina que sirve de transporte hacia la matriz de la mitocondria. Cualquiera que sea el nutrimento inicial, el producto final y común del metabolismo es el ácido acético, cuyo radical acetil se combina con la coenzima A.
En la segunda etapa, que se desarrolla igualmente en la cámara interna o matriz de la mitocondria, la acetilcoenzima A cede el radical acetil (CH3CO) al ácido oxalacético y entra en el ciclo de Krebs, que es una sucesión de reacciones oxidativas transformando cada radical acetil en 2 moléculas de CO2, 8 átomos de hidrógeno y 8 electrones. Los electrones son inmediatamente captados por la nicotamida adenina dinucleótido (NAD) y por la flavina adenina dinucleótido (FAD) para ser transmitidos sobre el aceptor final, en este caso el oxígeno molecular para un esamblaje de complejos asociados en una cadena respiratoria. El rendimiento del ciclo tricarboxílico es muy importante, puesto que una molécula de glucosa permite la obtención de 38 moléculas de ATP, en tanto que la glucólisis en anaerobiosis no suministra más que dos.
En la cadena respiratoria, tercera etapa de la producción de energía localizada en la membrana interna, los complejos siguientes van a comenzar a actuar:
Complejo I: NADH-CoQ reductasa (cerca de 40 subunidades).
Complejo II: succinato-CoQ reductasa (4 subunidades).
Complejo III: ubiquinona-citocromo C reductasa (11 subunidades).
Complejo IV: citocromo C oxidasa (13 subunidades).
El complejo V o ATPasa (14 subunidades) asegura la síntesis de ATP a partir del ADP y del folato inorgánico en la matriz mitocondrial1,4 (fig. 1)10.
Muchas enfermedades se han clasificado de forma provisional como trastornos mitocondriales debido a una forma o una bioquímica anormal de las mitocondrias, o bien a un modo de herencia materna. Las mutaciones de ADNmt asociadas a la enfermedad constituyen en la actualidad un criterio diagnóstico esencial. Aunque cada fenotipo de la enfermedad mitocondrial posee rasgos clínicos deferenciados (tabla II)9, todos ellos comparten distintas características clínicas y de laboratorio (tablas III y IV)9.



Neuropatía óptica hereditaria de Leber (NOHL)
Esta enfermedad se manifiesta por la pérdida bilateral de la visión, subaguda e indolora, con escotoma central y discromatopsia. Esta neuropatía óptica es hereditaria y se conoce como enfermedad de Leber (NOHL) y se caracteriza por la pérdida rápida de la visión central durante la edad adulta. Los ojos pueden ser afectados en forma simultánea o en forma secuencial. La edad media de aparición es de 23 años y los varones se afectan 3 o 4 veces más que las mujeres. El fenotipo de la neuropatía óptica hereditaria de Leber apenas se asemeja clínicamente al de otras enfermedades mitocondriales. En un primer momento se asoció con este grupo debido al modo de herencia materno. Se ha informado que las pacientes con NOHL y sus familiares maternos también manifiestan una variedad de síntomas adicionales. Se han observado defectos de la conducción cardíaca en algunas familias28. Se han descrito problemas neurológicos leves, tales como reflejos alterados, ataxia y una neuropatía sensorial, así como anormalidades esqueléticas. En los varones que heredan la mutación en el par de bases (pb) 11778, la penetrancia es de aproximadamente el 50%, pero sólo del 20% en las mujeres. Esta diferencia no se puede explicar por una heteroplasmia y se cree que sugiere el compromiso de un gen ligado al cromosoma X que puede ser afectado por una variedad de mutaciones29.
La fisiopatología de la pérdida de visión se basa en factores genéticos y epigenéticos (tabaco y alcohol). Las mutaciones del ADNmt muestran gran heterogeneidad genética. Las principales mutaciones del ADNmt asociadas a NOHL afectan sobre todo a los genes del complejo I (en las posiciones nucleotídicas 11778 (gen ND-4), 3460 (gen ND-1) y 14484 (gen ND-6) (fig. 1)10. Existen otras mutaciones del ADNmt que también intervienen en el NOHL, aunque en menor medida, como una mutación del nucleótido 13708 (gen ND-5) del ADNmt del halogrupo J que se da en la raza blanca.
Se están descubriendo correlaciones entre el genotipo y el fenotipo para las principales mutaciones del ADNmt asociadas a NOHL. Por ejemplo, el pronóstico de la recuperación de la visión varía unas 10 veces dependiendo de la mutación. También se han descrito mutaciones del ADNmt que provocan NOHL y dis-tonía9.
Síndrome de mioclonía, epilepsia y fibras rojas melladas (MERRF)
Llamado también epilepsia mioclónica con fibras rojas y rotas9. El término fibras rojas melladas deriva de las características histológicas observadas con la tinción tricrómica de Gomota modificada en el múscu lo fresco congelado, en el cual las mitocondrias acumuladas se ven rojas como resultado de reordenamientos del ADNmt o mutaciones puntuales que afectan a genes del ARNt.
El síndrome MERRF comprende mioclonía, convulsiones generalizadas, miopatía mitocondrial, ataxia cerebelosa, pérdida de la audición y deterioro intelectual30. Una mutación A a G en los nucleótidos 8344 y 8356 del gen ARNmt L y S es causante del 80 al 90% de los casos del síndrome de MERRF. La mutación es de cambio de sentido en el gen de ARN de transferencia de la lisina que produce múltiples deficiencias en los complejos enzimáticos de la cadena respiratoria26. El fenotipo clínico varía enormemente en el mismo linaje, el cual es compatible con una población heteroplásmica de ADNmt.
Son característicos los rasgos neurológicos y de laboratorio comunes a otras encefalomiopatías mitocondriales. Los parientes maternos permanecen asintomáticos o presentan síndromes clínicos parciales, incluidos lipomas en una distribución característica en forma de «collera» e hipertensión10.
Encefalomielopatía mitocondrial, acidosis láctica y episodios afines al ictus (MELAS)
Este síndrome consiste en encefalomielopatía, acidosis láctica y episodios similares a accidentes cerebrovasculares mitocondriales (MEALESACVM). Los acontecimientos similares al ictus provocan disfunción cerebral subaguda, lesiones de la estructura cerebral, crisis convulsivas y otros rasgos clínicos y de laboratorio comunes (tablas III y IV)9. La herencia materna del síndrome MELAS quizá quede eclipsada por los rasgos clínicos leves que presentan los miembros de la familia. Una mutación puntual del nucleótido 3243 del gen ARNt leu (URR) provoca el 80% de los casos de MELAS. No obstante, las características clínicas de la mutación del ADNmt en el nucleótido 3243 son polimorfas; este síndrome se asocia además con oftalmplejía externa progresiva crónica (OEPC) sin deleción, miopatía, sordera, diabetes y distonía9.
Se manifiesta por primera vez durante la niñez como un crecimiento detenido, episodios similares a accidentes cerebrovasculares recurrentes que se manifiestan como hemiparesia, hemiapnosia y ceguera cortical. También puede haber convulsiones focales o generalizadas, epilepsia mioclónica y pérdida de la audición, y en ocasiones se observan vómitos episódicos. La muerte a menudo ocurre antes de los 20 años de edad. Como hemos señalado, esta enfermedad se asocia con una mutación puntual en ARNt de la leu-cina31.
Síndrome de Kearns-Sayre con oftalmoplejía externa progresiva crónica
El síndrome de Kearns-Sayre con oftalmoplejía externa progresiva crónica (SKS/OEPC) se caracteriza por oftalmoplejía, ptosis, retinitis pigmentaria atípica y miopatía mitocondrial. A veces se observan rasgos clínicos adicionales (tabla III)9, además de las anomalías de laboratorio características de los trastornos mitocondriales (tabla IV)9. Los pacientes con OEPC presentan irregularidades en la biopsia del músculo esquelético, fibras rojas y rotas y cambios ultraestructurales. Cuando aparecen hallazgos somáticos y del sistema nervioso central junto con la oftalmoplejía externa progresiva crónica, reciben el nombre de síndrome «OEPC plus». El síndrome de Kearns-Saye, para algunos autores, es un subgrupo del OEPC plus que comienza antes de los 20 años y se caracteriza por OEPC y retinopatía pigmentaria atípica; otros rasgos comprenden elevación de las proteínas del líquido cefalorraquídeo, ataxia y bloqueo cardíaco (defectos de la conducción cardíaca)10,30.
Otros aspectos pueden incluir pérdida de la audición, demencia, baja estatura, caracteres sexuales secundarios retardados, hipoparatiroidismo e hipotiroidismo30.
Casi todos los enfermos con OEPC portan una deleción única y grande de ADNmt que puede detectarse con precisión mediante los métodos de genética molecular. No obstante, el carácter esporádico de casi todas las deleciones únicas del ADNmt obliga a extraerlo del músculo esquelético. La retinitis pigmentosa es un claro factor predictivo de la deleción. Aún no se conoce el mecanismo por el que se producen deleciones de ADN, aunque el más verosímil parece la recombinación o el desapareamiento durante la replicación. Las uniones de casi todas las deleciones contienen secuencias repetidas directamente, incluido «un lugar favorito» con repetición directa de 13 nucleótidos, que justifica alrededor del 25% de las deleciones. Casi la mitad de todas las eliminaciones están delimitadas por otras repeticiones directas y la cuarta parte carece de ellas. Algunas pacientes presentan una duplicación parcial de las moléculas de ADNmt. Se ha hallado una mutación puntual del nucleótido 3243 en muchos enfermos que carecen de eliminaciones y duplicaciones del ADNmt10. Según Kramer et al30, las mutaciones de ADNmt, más comúnmente deleciones, en general ocurren de forma espontánea, por ende esta enfermedad no es hereditaria. Habitualmente se demuestra heteroplasmia en el ADNmt muscular30.
Neuropatía, ataxia y retinitis pigmentosa/enfermedad de Leigh heredada de la madre
Como su nombre indica, esta enfermedad mitocondrial consiste en neuropatía, ataxia y retinitis pigmentosa (NARP), y se caracteriza por una combinación variable de debilidad proximal, neuropatía sensorial, retraso del desarrollo, ataxia, crisis convulsivas, demencia y degeneración pigmentaria de la retina. Esta enfermedad fue descrita por primera vez en 1990 por Holt et al32 y se asocia con una mutación puntual en el ADNmt en el gen para la subunidad 6 de la H+-ATPasa mitocondrial. Esta misma mutación se ha observado en familias con la enfermedad de Leigh (ver más adelante)30.
Este trastorno heredado de la madre se asocia a 2 mutaciones heteroplásmicas de sentido erróneo del nucleótido 8993 del gen ATPasa 6, ya citado.
Deficiencia de citocromo C oxidasa (deficiencia de complejo IV)
Hay 3 síndromes clínicos establecidos con deficiencia de citocromo C oxidasa (COX), dos de los cuales representan formas variantes de una miopatía infantil. Una de estas formas es benigna y se caracteriza por presentar una recuperación espontánea hacia los 2 o 3 años de edad. La otra forma se presenta durante el período neonatal y da como resultado una insuficiencia respiratoria. La forma fatal también se asocia con un defecto tubular renal, se hereda como un rasgo recesivo y se cree que representa un defecto en un polipéptido codificado por un gen nuclear de la cadena respiratoria.
La tercera forma de deficiencia de COX afecta al sistema nervioso central y se conoce como síndrome de Leigh (ya aludido antes), de herencia materna. La enfermedad de Leigh de carácter autosómico recesivo se asocia con déficit de citocromo C oxidasa y está causada por un déficit de la proteína codificada en el núcleo, SURF1, necesaria para la biogénesis del complejo citocromo C oxidasa (complejo IV). Las mutaciones puntuales del ADNmt asociadas con los síndromes NARP, MELAS y MERRF y con otros trastornos mitocondriales se detectan con facilidad mediante análisis de genética molecular del ADNmt extraído del músculo o la sangre.
La presentación típica del síndrome de Leigh durante el período neonatal consiste en hipotonía, vómitos y una retinitis pigmentaria que lleva a la pérdida de visión. Las concentraciones de ácido láctico están aumentadas en la sangre y el líquido cefalorraquídeo. Se han demostrado diversas mutaciones heteroplásmicas en el ADNmt de los pacientes con el síndrome de Leigh30.
Diabetes mellitus heredada por vía materna
En diversos estudios retrospectivos se demostró que los pacientes con diabetes mellitus no dependiente de insulina era mucho más probable que tuvieran una madre con el mismo diagnóstico y no el padre33. En un estudio se observó que esto también era cierto en el caso de las mujeres con diabetes gestacional34. Sin embargo, estos estudios pueden estar sujetos a ciertas desviaciones porque en ellos se utilizó la información de los pacientes para comprobar la existencia de familiares de primer grado afectados.
Se ha informado de intolerancia a la glucosa o diabetes mellitus no dependiente de insulina en algunos pacientes con miopatía mitocondrial. Hasta el 2% de los pacientes con MEALESACVM son diabéticos. La diabetes de este grupo de pacientes se ha asociado con sordera nerviosa y una mutación puntual en un gen mitocondrial para el ARNt de la leucina35. Por lo tanto, parece probable que existan mutaciones mitocondriales involucradas en la patogenia de una proporción pequeña, pero clínicamente significativa, de casos de diabetes mellitus no dependiente de insu-lina30.
Múltiples deleciones del ADN mitocondrial transmitidas de forma autosómica
Se ha averiguado que diversas familias con variantes clínicas de la OEPC albergan múltiples deleciones del ADNmt del músculo esquelético. La herencia autosómica de múltiples deleciones del ADNmt implica un defecto primario de un gen del ADN nuclear que repercute de forma secundaria en la calidad del ADNmt. Las múltiples deleciones del ADNmt pueden transmitirse con carácter autosómico dominante o autosómico recesivo, aunque a veces adoptan la forma de mutaciones somáticas9.
La timidita fosforilasa fue el primer gran gen nuclear descubierto con influencia del tipo «trans» en la regulación y en la función del ADNmt normal. Las mutaciones de la timidita fosforilasa provocan una enfermedad autosómica recesiva denominada «encefalomiopatía mioneurogastrointestinal» (EMNGI). Se han relacionado diversos loci nucleares en las formas autosómicas dominantes de la OEPC. La pérdida de ADNmt específica de los tejidos y transmitida con carácter autosómico representa un defecto cuantitativo del ADNmt causado por un error de comunicación intergenómica9,10,36.
Miopatía mitocondrial
La miopatía mitocondrial se caracteriza por debilidad proximal fija acompañada de una notable intolerancia al esfuerzo. Los individuos afectados por miopatía mitocondrial manifiestan debilidad, intolerancia al ejercicio y mioglobinuria después del ejercicio prolongado. Las miopatías mitocondriales se caracterizan por defectos en las vías de producción de energía de las mitocondrias. Existen 4 tipos principales clasificados por el sitio específico del defecto: defectos en el transporte de sustratos, defectos en la cadena respiratoria, defectos en la utilización de sustratos y defectos en la conservación y utilización de energía. Sólo existen presentaciones de casos en los 2 primeros tipos de defectos en las mujeres embarazadas37.
Un tipo de defecto en el transporte de sustratos comprende una deficiencia de carnitina. La deficiencia sistémica de carnitina es un trastorno raro; la deficiencia secundaria de carnitina en el embarazo es más frecuente38. Se trata de un trastorno potencialmente fatal que puede ser exacerbado por el transporte placentario de carnitina y se ha tratado con éxito con suplemento de carnitina. La dosificación recomendada en estas mujeres fue de 2 g diarios39.
Otro defecto del transporte de sustratos involucra a la enzima carnitina palmitoiltransferasa (CPT). Esta enzima es necesaria para el transporte de ácidos grasos a través de la membrana mitocondrial interna. Se ha comunicado un caso de deficiencia de carnitina palmitoiltransferasa en el embarazo15. La paciente tenía valores bajos de CPT en las biopsias del músculo estriado y liso. Se le practicó una intervención cesárea por falta de progresión durante su primer parto y tuvo un parto vaginal con éxito en un embarazo ulterior. No experimentó debilidad muscular posparto ni intolerancia al ejercicio.
Hasta 1990, hay un único caso comunicado de embarazo en una mujer con un defecto en la cadena respiratoria40. Después se han publicado, como veremos, más casos, además del aquí reflejado. Aquella mujer experimentó intolerancia leve al ejercicio, pero su embarazo y parto no tuvieron complicaciones40. La paciente no presentó exacerbación de sus síntomas después del parto.
En general, como señalamos, los pacientes con miopatía mitocondrial presentan fatiga y falta de resistencia, aunque la etiología mitocondrial a menudo se contempla junto con otros rasgos neurológicos, somáticos o de laboratorio. La rabdomiólisis franca es rara. En la electromiografía se observa una miopa tía no irritativa, y la creatincinasa sérica suele encontrarse normal o ligeramente elevada. La biopsia del músculo esquelético muestra la proliferación anómala de mitocondrias y «fibras rojas y rotas», una característica histológica de los graves defectos bioquímicos de la fosforilación oxidativa. En la miopatía mitocondrial son típicas las grandes deleciones y una variedad de mutaciones puntuales del ADNmt9,10.
El asesoramiento genético es importante en las miopatías mitocondriales. Dado que sólo el ovocito contribuye con las mitocondrias y por lo tanto con el ADN mitocondrial para el nuevo cigoto, se observa un patrón de herencia puramente materno, dato ya reflejado en este estudio más atrás.
Enfoque de los signos clínicos
Los signos clínicos de la citopatía mitocondrial aparecen preferentemente en los órganos que poseen un gran consumo de energía como el cerebro, los riñones, el corazón, los músculos y el hígado. Johns10, gráficamente, comenta que casi todos los tejidos del organismo dependen, en cierta medida, del metabolismo oxidativo, por lo que los pacientes con enfermedad mitocondrial pueden acudir a muchos médicos especialistas. Las manifestaciones somáticas que se enumeran en la tabla III9, observadas en un primer momento en asociación con las enfermedades mitocondriales clásicas, pueden ser el síntoma clínico habitual o dominante o ser rasgos comórbidos importantes.
Existen formas dramáticas que se manifiestan desde el nacimiento por un coma cetoacidósico con convulsiones, una insuficiencia hepática, una tubulopatía proximal, una miocardiopatía o un síndrome de Pearson (anemia sideroblástica con neutropenia, trombopenia, insuficiencia hepática, atrofia vellosa, insuficiencia pancreática externa).
Otras formas aparecen más tarde en la infancia o en la edad adulta (tabla V).

Los primeros signos son, con frecuencia, una debilidad muscular, una intolerancia al esfuerzo y una oftalmoplejía externa progresiva. Las manifestaciones oftalmológicas de las mutaciones del ADNmt son notables y afectan prácticamente a todo el eje visual, desde los párpados, la córnea y los músculos extraoculares hasta la corteza occipital. Los principales hallazgos oculares comprenden oftalmoplejía, neuropatía óptica y retinopatía pigmentaria.
Ciertas asociaciones clínicas se han descrito y reagrupado en grandes síndromes, y ya se han aclarado sus formas de transmisión14.
El fenotipo de la enfermedad mitocondrial es diferente según que la mutación aparezca precozmente, en el estadio de ovocito o de blastocisto, con una afección multifocal (síndrome de Kearns-Sayre), o si aparece después de la diferenciación celular, con una expresión más localizada (afección ocular o del músculo esquelético)16. Un mismo cuadro clínico puede corresponder a transmisiones hereditarias diferentes (síndrome de Leigh)41.
Desde el punto de vista biológico, la perturbación de la cascada de las reacciones de oxidorreducción y la acumulación de los productos sintetizados «río arriba» entrañan un aumento secundario de la concentración de los lactatos en el citoplasma, después en el plasma, a veces evidentes en reposo, pero más frecuentemente después de un esfuerzo.
La biopsia muscular muestra fibras musculares hechas jirones, dilaceradas por la acumulación periférica y entre las miofibrillas, de mitocondrias anormales, evidenciadas por coloración tricroma de Gomori modificada16.
El diagnóstico bioquímico del déficit de funcionamiento de la cadena respiratoria es esencial y se realiza preferentemente en los tejidos clínicamente afectados (músculo, linfocitos). No obstante, cualquiera que sea el órgano afectado, es esencial llevar a cabo una biopsia de la piel en función de futuras investigaciones enzimológicas o moleculares en los fibroblastos en cultivo para permitir un eventual diagnóstico prenatal en ulteriores gestaciones. La polarografía mide el consumo de oxígeno en fracciones enriquecidas en mitocondrias con la ayuda de un electrodo de Clark en presencia de diferentes sustratos. Este método no puede efectuarse más que en material fresco. La espectrofotometría mide las actividades de los complejos enzimáticos en solitario o en grupo, utilizando donantes o receptores específicos de electrones. Esta técnica se realiza a partir de biopsias de pequeña «talla» que deben ser inmediatamente congeladas y mantenidas permanentemente en nitrógeno líquido14.
De esta forma el estudio bioquímico permite obtener el diagnóstico del déficit de la cadena respiratoria. Los déficit pueden afectar a todos los complejos, de forma aislada o reagrupada. El porcentaje de los complejos afectados y por orden decreciente son: complejo I (33%), complejo IV (28%), complejo I+IV (28%), complejo III (7%) y complejo II (4%)14. No hay ninguna correlación entre el fenotipo bioquímico y el fenotipo clínico.
Diagnóstico molecular: ya hemos señalado con anterioridad la pluralidad de la transmisión genética. En efecto, un déficit de la fosforilación oxidativa puede tener un origen mitocondrial o un origen nuclear. Todos los modos posibles de transmisión ya se han señalado: exclusivamente materna, esporádica, autosómica dominante, autosómica recesiva, ligada al X42,43. Es útil la realización de un árbol genealógico detallado que permitirá entonces presagiar el modo de transmisión. Así, una transmisión únicamente materna evoca que en ésta hay una mutación del ADN mitocondrial.
Retomando el tema de los signos y síntomas clínicos, señalemos que las manifestaciones cardiovasculares consisten en miocardiopatía dilatada e hipertrófica, enfermedad de conducción y bloqueo cardíaco, síndrome de Wolf-Parkinson-White e hipertensión. La prevalencia de diabetes mellitus es mayor de la esperada en los pacientes con encefalomiopatías mitocondriales y aparece asociada a diversas mutaciones del ADNmt. Esta enfermedad se ha relacionado con la mutación puntual del nucleótido 3243, a menudo aunque no siempre unido a la sordera neurosensitiva44.
Enfermedad mitocondrial y gestación. Las alteraciones del ADNmt
El ADN mitocondrial, pequeño genoma circular de 16.569 pares de bases, está codificado por 37 genes: el ARN ribosomal, el ARN de transferencia y las 13 proteínas incluidas en la cadena respiratoria. El ADNmt es transmitido únicamente por la madre, puesto que el espermatozoide pierde todas sus mitocondrias citoplásmicas antes de la fecundación. El ADNmt parece ser susceptible de mutaciones de novo: deleción, inserción, sustitución.
Se han documentado ciertas mutaciones puntuales en casos de citopatías mitocondriales de transmisión materna, por ejemplo la atrofia óptica de Leber afectando al complejo I. En los síndromes MELAS y MERF, las principales mutaciones del ARN de transferencia se han encontrado en posición A3243G y G8344A2, respectivamente.
La severidad del cuadro clínico y la multiplicidad de los órganos afectados depende menos de la amplitud de la deleción que de la diseminación de las mutaciones del ADNmt en el organismo. Así la mutación sobre el nucleótido T8993G es superior al 90% en el síndrome de Leigh e intermedio, entre el 60 y el 80%, en el síndrome NRAP (debilidad muscular neurógena, ataxia y retinitis pigmentaria)45.
En la oftalmoplejía progresiva con miopatía proximal y en el síndrome de Kearns-Sayre una deleción común de gran amplitud (5.000 pares de bases) se ha evidenciado en casos esporádicos16.
Las alteraciones del genoma nuclear: los genes nucleares codifican para la mayor parte de las proteínas de la cadena respiratoria y siguiendo las leyes de la genética mendeliana. Aunque estos genes sean conocidos, solamente algunos de ellos han sido realmente considerados como causa de la enfermedad (citocromo oxidasa, NAD deshidrogenasa, succinato deshidrogenasa), lo que limita el alcance actual del diagnóstico prenatal molecular3,46,47. Existe en estas familias una tasa elevada de consanguinidad48.
En el caso específico de nuestra paciente, no se pudo encontrar la mutación genética, por lo que el diagnóstico se basó en los análisis polarográficos y espectrofotométricos y en la biopsia muscular.
Los primeros casos de citopatías mitocondriales asociados a la gestación se han descrito en mujeres que presentaban una anomalía de la beta-oxidación de los ácidos grasos, seguida de un déficit en la carnitina palmitoiltransferasa o en 3 hidroxiacil-CoA deshidrogenasa15,49. Estas gestaciones estaban complicadas por cardiomiopatía y rabdomiólisis, colapso en el posparto e insuficiencia renal en 2 casos sobre 3. Hemos encontrado una última referencia sobre citopatía mitocondrial y gestación, descrita por Racine et al50.
Por el contrario, cuando se trata de un desorden de la cadena respiratoria, la evolución de la gestación puede ser del todo satisfactoria, como en el síndrome de Kearns-Sayre16. Debemos, pues, distinguir las consecuencias de las mitocondriopatías en el embarazo y, en un segundo término, las de la gestación en la enfermedad materna.
El riesgo principal involucrado en la evolución de la gestación parece ser la hipertensión arterial gravídica y la preeclampsia17,51,52. En 2 familias que presentaron una gran incidencia de toxemias y de eclampsias, Folgero et al53 pusieron en evidencia dos mutaciones del ARN de transferencia, en las pacientes, en A3243G y en A12308G. En la observación de Racine et al50, con bastantes similitudes en nuestro caso, la toxemia se estableció en una afección renal preexistente. En caso de complicaciones hipertensivas, el tratamiento instaurado debe evitar el sulfato de magnesio que modifica el transporte del calcio, dado que la cascada de reacciones mitocondriales es dependiente del calcio54.
En la observación de Racine et al50, se objetivó un retardo de crecimiento intrauterino en relación con las anomalías de la velocimetría Doppler uterina. Ewart et al17 comunicaron un paro en el crecimiento fetal a las 32 semanas de amenorrea, pero los elementos Doppler no eran lo suficientemente peyorativos como para motivar la extracción fetal, la cual tuvo que efectuarse en el caso descrito por Racine et al50, y en nuestra observación.
Teniendo en cuenta la gravedad de la preeclampsia y de la insuficiencia renal, Racine et al50 fueron partidarios de extraer prematuramente el feto. Se han comunicado y comentado en la bibliografía otros 2 nacimientos prematuros espontáneos, a las 3517 y 3618 semanas de amenorrea.
La asociación de la enfermedad mitocondrial a una diabetes apareció en el caso de Racine et al50 desde la semana 26 de amenorrea, hecho igualmente reflejado en su observación por Kovilam et al18. Una mutación del ADNmt causante de un subtipo de diabetes tipo I asociada a una sordera se ha relacionado con una transposición A3243G en el genoma mitocondrial18: se trata de hecho del mismo gen que el que se ha encontrado mutado en el síndrome MELAS55,56.
La mayor parte de las pacientes en los trabajos publicados han parido por las vías naturales, algunas de ellas con ayuda instrumental en el momento de la extracción fetal por defecto de los esfuerzos del expulsivo. Blake et al16 opinan que las citopatías mitocondriales engendrarían un trabajo de parto más largo, con esfuerzos expulsivos poco eficaces. En realidad la ayuda en el expulsivo está, a menudo, sugerida para no favorecer un incremento de la acidosis por la producción de lactatos sanguíneos consecutiva a la actividad muscular y para evitar una rabdomiólisis en caso de déficit de 3 hidroxiacil-CoA deshidrogenasa o en carnitina palmitoiltransferasa15,49. En esta última situación, la afección de las fibras musculares lisas ha sido puesta en evidencia tras biopsias del miometrio en el curso de la cesárea15. Racine et al50 recuerdan que su paciente tuvo 2 gestaciones extrauterinas sin factor presdisponente evidente, fuera de la eventual debilidad contráctil de la musculatura tubular.
El parto por vías naturales está aconsejado, en ausencia de complicación grave de la gestación. Es preciso privilegiar la analgesia locorregional para disminuir los esfuerzos del estrés y evitar la acidosis láctica materna. En caso de anestesia general, es preciso evitar los fármacos miorrelajantes16. En cuanto al posparto, se han descrito algunas complicaciones en la bibliografía: Soccio et al21 han publicado el caso de una paciente que tuvo hemorragia de media intensidad en el alumbramiento, resuelta con tratamiento adecuado, y Kovilam et al18 han comunicado alteraciones del ritmo cardíaco en consonancia con una cardiomiopatía posparto.
Hay escasos datos que comuniquen las consecuencias de la gestación en la enfermedad mitocondrial. En la publicación de Soccio et al21, la paciente afectada de un déficit de citocromo C oxidasa presentó durante su embarazo una astenia progresiva, provocada por una infección de vías respiratorias en la semana 20 de amenorrea, y fue necesaria una inmovilización prolongada hasta el fin de su gestación. Da fe de la gravedad de la sintomatología de la mitocondriopatía el hecho de que fuera preciso una cesárea para la extracción fetal21. Racine et al50 constatan que los síntomas neurológicos y oculares permanecieron estables durante todo el embarazo. Por el contrario, la insuficiencia renal experimentó un notable agravamiento, lo que precipitó la extracción prematura por cesárea50, al igual que en nuestro caso. En el posparto, las anomalías del funcionamiento renal mejoran parcialmente, pero la paciente ha quedado con insuficiencia renal terminal, con la idea de que será necesario un trasplante50.
La gestación puede ser igualmente reveladora de una afección mitocondrial latente, como se puede observar en los casos documentados por Kokawa et al19 y Yanagawa et al20, en que se diagnosticaron síndromes MELAS en el tercer trimestre de la gestación. Los embarazos y los partos se desarrollaron normalmente después de administrar tratamientos apropiados, como coenzima Q10 y dicloroacetato.
El diagnóstico prenatal (DAN) debe tener en cuenta el polimorfismo de la enfermedad. No es en realidad factible más que en el caso de que exista un caso índice identificador en la familia57, pues el diagnóstico descansa como mínimo en el estudio de los fibroblastos cutáneos e, idealmente, en el descubrimiento de una mutación. El diagnóstico bioquímico utiliza la polarografía y la espectrofotometría sobre las vellosidades coriales entre las semanas 9 y la 11 de amenorrea, o en la amniocentesis hacia la semana 16 de amenorrea, pues estas células tienen un origen embriológico común con los fibroblastos58. No obstante, solamente el 50% de los sujetos afectados expresan su déficit enzimático en cultivos celulares de fibroblastos de la piel46. Por otra parte, persisten numerosas incógnitas sobre la cinética del ensamblaje de los complejos mitocondriales durante la vida embrionaria, después fetal. Sería necesario, pues, confirmar una ausencia de alteración en el trofoblasto por un segundo estudio posterior sobre las células amnióticas, hacia la semana 28 de amenorrea. Por otra parte, la actividad del complejo I es entorpecida por la fuer te actividad de la NADH reductasa no mitocondrial46. Es del todo evidente que es preciso privilegiar la búsqueda molecular de una mutación, puesto que se conoce mucho antes que la expresión enzimática del gen59.
El ADN molecular después de biopsia trofoblástica o amniocentesis aparece por consecuencia preferiblemente cuando la mutación de la cadena respiratoria del gen es conocida. No obstante, para evitar errores de diagnóstico por exceso o por defecto, importa excluir una contaminación materna de la toma y buscar la contribución paterna del genoma47,60. Las modificaciones de gran tamaño del ADNmt se encuentran, en la gran mayoría de los casos, de manera esporádica61. Las mutaciones puntuales del ADNmt, casi siempre en el estado heteroplásmico, se transmiten exclusivamente según un modo materno62. No obstante, es muy difícil pronunciarse sobre el riesgo del niño, pues la proporción de moléculas del ADNmt en las vellosidades coriales no permite estimar su importancia en otros tejidos fetales ni su evolución en el curso del desarrollo embrionario14,63.
La gestación instalada en una paciente afectada de citopatía mitocondrial es un embarazo de alto riesgo. Se debe efectuar las consultas con los especialistas en función de los órganos dañados. Una atenta lectura del examen biológico de las trisomías (ß-HCG) y la práctica de una velocitometría de las arterias uterinas pueden identificar el riesgo de preeclampsia. Se debe realizar la búsqueda sistemática de una diabetes gestacional en el primer trimestre. El examen morfológico del feto por ecografía y mediante resonancia magnética nuclear debe excluir anomalías cerebrales en casos de piruvato deshidrogenasa (ventriculomegalia y calcificaciones intracerebrales)64 o de afecciones de los complejos III y IV de la cadena respiratoria (hidrocefalia e hidranencefalia)65. Si es posible, se debe identificar el ADN. El parto por vía vaginal puede ser viable, de preferencia con analgesia locorregional para eliminar el estrés y eludir así la acidosis láctica; las alteraciones neurológicas no parecen ser una contraindicación a la analgesia epidural66,67.
Los medios terapéuticos específicos para el tratamiento de las mitocondriopatías son muy limitados y tienen una acción esencialmente sintomática. Las perfusiones de bicarbonato tratan de impedir la acidosis láctica; la diálisis puede mejorar temporalmente la insuficiencia renal. La coenzima Q es activa, sobre todo en las formas musculares. En la bibliografía se han encontrado casos de mujeres gestantes con esta prescripción, pero el efecto de este medicamento en la gestación no está en ningún caso bien documentado. Se han propuesto otros tratamientos paliativos: la creatinina, el dicloroacetato, los antioxidantes como el ácido ascórbico y la vitamina E, vitaminas como la tiamina o la riboflavina, sustratos de la fosforilación oxidativa como el succinato o la carnitina68-71.
El consejo genético es esencial para evaluar el riesgo de transmisión. Éste es inferior al 5% en el caso de oftalmoplejía externa progresiva o del síndrome de Kearns-Sayre. Por el contrario, en otros síndromes, el riesgo es proporcional a la carga mutante, es decir, al porcentaje de ADNmt mutados en los linfocitos de la sangre: el 20% de riesgo si la carga está entre el 1 y el 19% y del orden del 50% si ésta es superior o igual al 25% en el síndrome MELAS con la mutación A3243G, puesto que el riesgo es estimado inferior al 5% si la carga mutante está por debajo del 35% en el síndrome MELAS con la mutación G83344A. En la atrofia óptica de Leber, el riesgo es superior al 30% en caso de hermanos, e igual al 8% en caso de hermanas, del 46% en los sobrinos, el 10% en caso de las sobrinas, el 31% en el caso de los primos y el 6% en el caso de las primas2. En caso de riesgo elevado, se podría tener en cuenta un diagnóstico preimplantatorio o una donación de ovocitos en caso de transmisión materna43,44,64.
Referente a los aspectos anestésicos, y en concreto en casos de miopatía mitocondrial, Crochetiére72 señala que dado que tanto la deficiencia de carnitina palmitoiltransferasa como la hipertermia maligna pueden ocasionar rabdomiólisis, algunos especialistas creen que existe una relación entre ambas73. Los facultativos pueden considerar la posibilidad de hipertermia maligna, la omisión de los fármacos desencadenantes y la vigilancia de la temperatura72.
Importancia de las mutaciones del ADN mitocondrial en las enfermedades prevalentes
La importancia de las mutaciones del ADNmt en enfermedades frecuentes y con repercusiones socioeconómicas está siendo objeto de una investigación ex haustiva. La base genética de muchas enfermedades prevalentes es compleja y no sigue un sencillo patrón de herencia mendeliana de un solo gen. Las enfermedades mitocondriales, como neuropatía óptica hereditaria de Leber y sordera inducida por los aminoglucósidos, ilustran las posibles interacciones fisiopatológicas entre los factores genéticos y epigenéticos. Como resultados de estas interacciones, las mutaciones de ADNmt intervienen en algunas enfermedades frecuentes, como la diabetes mellitus, en la que no se ha demostrado un patrón de herencia materno9,10.
La acumulación hística de mutaciones somáticas (no heredadas) de ADNmt probablemente guarde relación con algunos trastornos degenerativos de aparición tardía, como la enfermedad de Alzheimer o la de Parkinson. Se ha demostrado, por ejemplo, que con la edad las mutaciones del ADNmt se acumulan en los tejidos, incluidos algunos tejidos posmitóticos o en los que se renuevan con lentitud. La lesión oxidativa, como sucede en los episodios repetidos de isquemia y repercusión, incrementa de forma notable la acumulación de mutaciones del ADNmt, que también resulta afectado por los factores ambientales. El fármaco antirretroviral azidotimidina elimina el ADNmt muscular y origina una miopatía mitocondrial adquirida. La disfunción mitocondrial acumulada y dependiente de la edad, mediada en cierto modo por la lesión oxidativa que experimentan el ADNmt y otras macromoléculas mitocondriales, contribuye en gran medida al envejecimiento.
El diagnóstico inequívoco de enfermedad mitocondrial con ayuda de los métodos de genética molecular es un registro indispensable para las personas que deban recibir consejo genético y, en última instancia, tratamiento9,10,74.
CONCLUSIÓN
En conclusión, las citopatías mitocondriales son enfermedades raras. La frecuencia de las encefalomiopatías de origen mitocondrial en consonancia con una alteración de fosforilación oxidativa se estima en 1/11.000 para Olfords et al75. Se trata de enfermedades complejas por la diversidad de los síntomas, por la dificultad de obtener la prueba diagnóstica y por las múltiples modalidades de la transmisión genética. Es preciso saber y pensar en una embarazada que presenta, por una parte, alteraciones musculares, en particular una fatigabilidad excesiva asociada a parálisis oculares o un descenso de la agudeza visual; por otra parte, diversas alteraciones de órganos, aparentemente sin relación entre ellos, que tienen un tanto por ciento de riesgos de ser alterados porque son grandes consumidores de energía (cerebro, riñón, corazón e hígado). La gestación que se instala con esta enfermedad preexistente no está contraindicada, pero no obstante demanda un seguimiento atento pluridisciplinario. Efectivamente, hemos visto descritas numerosas complicaciones obstétricas documentadas en la bibliografía y que los síntomas maternos se podrían agravar por la misma gestación. El caso clínico aportado en esta revisión de enfermedades mitocondriales y gestación confirma los datos comentados por otros autores, dado que nuestra paciente ha desarrollado una diabetes gestacional y una preeclampsia severa, que precisó la extracción fetal de urgencia en la semana 30 de amenorrea. Además, es preciso descartar que la función renal, ya alterada por una nefropatía preexistente, se deterioró considerablemente en el curso de esta gestación. Se debe formular a la pareja una información clara y precisa ante la dificultad de establecer un diagnóstico prenatal. Éste sólo es posible si un caso índice ha sido identificado en la familia, mediante un estudio enzimático o molecular con, no obstante, numerosas incertidumbres en cuanto a la «representatividad» del espécimen ovular para determinar la diseminación real de la enfermedad mitocondrial y a la gravedad de su expresión clínica en el organismo del niño al nacer76,77.
