






High Density Lipoproteins (HDL) are dysfunctional in hypercholesterolemia patients. The hypothesis was tested that nicotinamide (NAM) administration will influence HDL metabolism and reverse cholesterol transport from macrophages to the liver and feces in vivo (m-RCT) in a murine model of hypercholesterolemia.
MethodsApolipoprotein E-deficient (KOE) mice were challenged with a high-fat diet for 4 weeks. The effect of different doses of NAM on cholesterol metabolism, and the ability of HDL to promote m-RCT was assessed.
ResultsThe administration of NAM to KOE mice produced an increase (∼1.5-fold; P<0.05) in the plasma levels of cholesterol, which was mainly accounted for by the non-HDL fraction. NAM produced a [3H]-cholesterol plasma accumulation (∼1.5-fold) in the m-RCT setting. As revealed by kinetic analysis, the latter was mainly explained by an impaired clearance of circulating non-HDL (∼0.8-fold). The relative content of [3H]-tracer was lowered in the livers (∼0.6-fold) and feces (>0.5-fold) of NAM-treated mice. This finding was accompanied by a significant (or trend close to significance) up-regulation of the relative gene expression of Abcg5 and Abcg8 in the liver (Abcg5: 2.9-fold; P<0.05; Abcg8: 2.4-fold; P=0.06) and small intestine (Abcg5: 2.1-fold; P=0.15; Abcg8: 1.9-fold; P<0.05) of high-dose, NAM-treated mice.
ConclusionThe data from this study show that the administration of NAM to KOE mice impaired m-RCT in vivo. This finding was partly due to a defective hepatic clearance of plasma non-HDL.
Las lipoproteínas de alta densidad (HDL) son disfuncionales en los pacientes hipercolesterolémicos. Hipotetizamos que la administración de nicotinamida (NAM) influirá en el metabolismo de las HDL y el transporte reverso de colesterol desde macrófagos (m-TRC) a hígado y a heces in vivo en un modelo de hipercolesterolemia en ratones.
MétodosSe utilizaron ratones con deficiencia de apolipoproteína E (KOE) tratados con una dieta rica en grasas durante 4 semanas. Se evaluó el efecto de diferentes dosis de NAM sobre el metabolismo del colesterol y el m-TRC.
ResultadosLa administración de NAM a ratones KOE produjo una elevación (∼1,5 veces) en los niveles plasmáticos de colesterol, principalmente debido a un aumento de la fracción no HDL. En ratones tratados con NAM se observó una acumulación de [3H]-colesterol en plasma (∼1,5 veces) en el ensayo m-TRC, siendo ello debido a una disminución en el aclaramiento de no HDL (∼0,8 veces). Esto último también se acompañó de unos niveles relativos hepáticos y fecales de radiactividad disminuidos en estos ratones (hígado: ∼0,6 veces; heces: >0,5 veces) y elevados de forma significativa o cercana a la significación en la expresión génica en el hígado (Abcg5: 2,9 veces; p<0,05; Abcg8: 2,4 veces; p=0,06) e intestino delgado (Abcg5: 2,1 veces; p=0,15; Abcg8: 1,9 veces; p<0,05) de ratones KOE tratados con la dosis alta de NAM.
ConclusiónNuestros datos muestran que la administración de NAM a ratones KOE afectó el m-TRC in vivo. Este hallazgo se debió en parte a un aclaramiento hepático defectuoso de lipoproteínas no HDL.
Familial hypercholesterolemia is characterized by elevated circulating levels of non-HDL cholesterol, particularly LDL cholesterol, cholesterol deposition in the arterial wall, and an increased risk of premature cardiovascular disease (CVD).1 The removal of LDL from the circulation is reduced in hypercholesterolemic patients and, conceivably, LDL accumulation may disturb the metabolism and atheroprotective properties of HDL. Indeed, HDL are dysfunctional and, in particular, the macrophage-specific reverse cholesterol transport in vivo (m-RCT), one of the most important anti-atherogenic functions of HDL,2 may also be damped in these patients.3
Statins have been proven effective to reduce CVD-related CVD mortality and morbidity, though not completely.4 Thus,in recent years there has been increased focus on other pharmacological agents, including those targeting HDL metabolism as additional therapy onto statins to further reduce residual CVD risk.5 However, the recent failure of HDL-C-raising agents, including nicotinic acid (NA) and CETP inhibitors, has challenged the use of HDL cholesterol as a risk factor and therapeutic target,6–10 thereby supporting the notion that agents increasing HDL function, rather than its cholesterol cargo, should be more effective for the translation of its atheroprotective properties.
The m-RCT pathway is considered as one of the most important mechanisms of HDL-mediated atheroprotection in vivo11 and its pharmacological induction has been proposed as strategy to reduce residual CVD risk. The first step is cholesterol efflux from lipid-laden macrophages to plasma HDL, which is mainly mediated by two cholesterol transporter ATP-binding cassettes, (Abc)a1 and Abcg1. Although cholesterol efflux capacity has been recently identified as an independent predictor of incident cardiovascular events,12 to trace cholesterol from macrophages during its transit to HDL, liver, and ultimately to feces is required to estimate the whole m-RCT-itinerary.13 Following cholesterol loading and plasma remodeling of HDL, these lipoproteins may be taken up by the liver via the SR-BI receptor. Hepatic cholesterol cargo may be then excreted into bile to be eventually released into feces. This step is mainly regulated by the cholesterol transporters Abcg5/g8 in the liver and the small intestine.13 Also, hepatic cholesterol may also be converted into bile acid by either the cytochrome p450 enzyme cholesterol 7α-hydroxylase (Cyp7a1) or 27α-hydroxylase (Cyp27a1), to be eventually released into bile by the apical bile salt export pump (Abcb11).
The role of nicotinic acid (NA), a water-soluble vitamin B3 derivative and an HDL-raising drug,14,15 on this process is rather controversial.16–20 Furthermore, its clinical use has been hindered by unpleasant side effects (i.e., flushing), and replaced by better-tolerated interventions. Conversely, nicotinamide (NAM), despite sharing structural and nutritional similarities with NA, does not cause flushing, but has not been proven effective in dyslipidemia and/or favorably influence plasma levels of HDL. The latter might have limited the assessment of the potential contribution, if any, of NAM on m-RCT. Thus, we tested the hypothesis that the administration of NAM will influence HDL metabolism and m-RCT in a murine model of hypercholesterolemia.
Materials and methodsMice and dietsAll animal procedures were conducted in accordance with published regulations and reviewed and approved by the Institutional Animal Care Committee of the Institut d’Investigacions Biomèdiques Sant Pau. Male ApoE deficient (KOE) mice on a C57BL/6J background were from Jackson Laboratories (Bar Harbor, ME, USA; no. 002052). Animals had free access to food and water and were used after a minimum of 7 days acclimatization to the housing conditions. At 8-week-old, mice (n=18), weighing 22–25g, were challenged to a high-fat diet (Western-type diet TD88137, ENVIGO, containing 21% of fat – saturated fat/total fat ratio=0.64 and 0.2% cholesterol) and randomly distributed into three groups (n=6 mice each) depending on whether they received tap water non-supplemented (untreated mice), or supplemented with either 0.25% NAM and 1% NAM for 4 weeks. The groups compared had an equal distribution of characteristics. Such experimental design was repeated in all kinetic analysis performed in this study. The dose of NAM administered to mice via tap water (1%) was established according to palatability criteria. From a previous experiment, there was a dose-related decrease in water consumption for mice receiving doses of NAM higher than 1% (data not shown). All animals were kept in a temperature-controlled environment (20°C) and humidity (66%) with a 12-hour (h) light/dark cycle. At the end of the studies, mice were kept mice were kept on a 4-h food deprivation, euthanized, and exsanguinated by cardiac puncture. Blood was collected and serum was obtained by centrifugation. Food and water intake were monitored for 2 days before the animals were euthanized.
Plasma lipids and lipoprotein analysisPlasma lipid and lipoprotein analyses were performed enzymatically using commercial kits adapted to a COBAS c501 autoanalyzer (Roche Diagnostics). HDL cholesterol was measured in apoB-depleted plasma obtained after precipitation with phosphotungstic acid and magnesium ions (Roche Diagnostics). Non-HDL and HDL fractions were isolated by ultracentrifugation at 100,000×g for 24h at a density of 1.063g/mL and 1.210g/mL, respectively.21 Lipoprotein protein concentrations were determined by the BCA method (Pierce).
Measurement of m-RCT in vivo[3H]-cholesterol-labeled J774A.1 macrophages were prepared and intraperitoneally injected into mice as described previously.22 Mice were then individually housed in metabolic cages and stools collected over the next two days. Plasma radioactivity was determined at 48h by liquid scintillation counting. HDL-associated [3H]-cholesterol was measured after precipitation of apoB-containing lipoproteins with phosphotungstic reagent. At the end of the assay, mice were euthanized, and livers and feces collected. Liver and fecal lipids were extracted with isopropyl alcohol-hexane. The lipid layer was collected, evaporated and [3H]-cholesterol radioactivity measured by liquid scintillation counting. The [3H]-tracer detected in fecal biliary acids was determined in the remaining aqueous phase of fecal material extracts. The amount of [3H]-tracer was either expressed as a fraction of the injected dose or relative to untreated mice, which were taken as 100%, respectively.
In vivo non-HDL turnoverAutologous [3H]-cholesteryl oleoyl ether-labeled non-HDL lipoproteins (containing 2.5×105cpm) obtained from KOE mice were isolated and intravenously injected into each mouse.21,23 Serum was collected into tubes at 2, 4, 6, and 24h under isofluorane anesthesis. Serum decay curves for the tracer were normalized to radioactivity at the initial 2-min time-point after tracer injection. Fractional catabolic rates (FCR) were calculated from the area under the serum disappearance curves fitted to a nonlinear, two-phase exponential decay model.21 The non-HDL FCR values were used to calculate the non-HDL cholesterol (non-HDL-C) secretion rate using the formula [(pool size×FCR)/g]. Serum volume was estimated to be 4% of body weight. At the end of the experiment, livers and the rest of the body were collected and subjected to lipid extraction. Liver [3H]-tracer was expressed as percentage of injected dose was also determined.
Distribution of intragastrically-administered [3H]-cholesterol in miceMice were given an oral fat gavage (OFG) consisting of 20μCi [3H]-labeled cholesterol ([1α,2α(n)-3H]-cholesterol, Perkin Elmer) in 200μL of virgin olive oil. Mice were bled by cardiac puncture at the times indicated after OFG over a period of 24h. The radioactivity in total plasma and the non-HDL fraction, and liver were determined by scintillation counting.
[3H]-cholesterol efflux from cultured mouse macrophage foam cellsThe ability of cholesterol efflux mediated by either mouse plasma or HDL was evaluated from [3H]-cholesterol-loaded J774A.1 macrophages.22 Briefly, cellular cholesterol was labeled for 48h of incubation with 2μCi/well [1α,2α(n)-3H]-cholesterol (Perkin Elmer). [3H]-cholesterol-labeled cells were incubated in DMEM containing 5% mouse plasma or HDL from ApoB-containing lipoprotein-depleted plasma, respectively, at 37°C for 4h. The media and cell lysates were collected and analyzed for [3H]-radioactivity by liquid scintillation. Cholesterol efflux (%) was calculated as cpmmedium/(cpmcells+cpmmedium)×100.
Quantitative real-time RT-PCR analysesThe RNA from both liver and small intestine was isolated using TRIzol® reagent (invitrogen) following the manufacturer's protocol and purified using the RNeasy Plus Mini Kit (Qiagen). The integrities of the total RNA samples were determined using a Bioanalyzer. Total RNA was reverse-transcribed with random primers using Oligo (dT)23 and a mixture of dNTPs (Sigma), and M-ML reverse Transcriptase, RNase H Minus, and Point Mutant (Promega) to generate cDNA. cDNA was subjected to real-time PCR amplification using Taqman Master Mix (Applied Biosystems). Specific mouse Taqman probes (Applied Biosystems) were used for Apoa1 (Mm00437569_m1), Abca1 (Mm00442646_m1), Abcb11 (Mm00446241_m1), Abcg1 (Mm00437390_m1), Abcg5 (Mm00446241_m1), Abcg8 (Mm00445970_m1), Cyp7a1 (Mm00484152_m1), Cyp27a1 (Mm00470430_m1), Cyp7b1 (Mm00484157_m1), Hmgcr (Mm01282499_m1), Ldlr (Mm00440169_m1), Lrpap1 (Mm00660272_m1), Lrp1 (Mm00464608_m1), Scarb1 (Mm00450236_m1), Vldlr (Mm00443298_m1), and Actb (Mm00607939_s1) (reference gene) in mouse tissue gene expression analysis. Real-time PCR assays were performed on a C1000 Thermal Cycler coupled to a CFX96 Real-Time System (Bio-Rad Laboratories SA, Life Science Group). All analyses were performed in duplicate. The levels of the reference gene were not influenced by the treatment with NAM (data not shown). The relative mRNA expression levels were calculated using the 2−ΔΔCt method.
Statistical methodsData are shown as mean±SEM. Differences between mean values were assessed by either the nonparametric U Mann–Whitney test or the nonparametric Kruskal–Wallis test followed by Dunn's post-test, as appropriate. Statistical significance was defined as P<0.05.
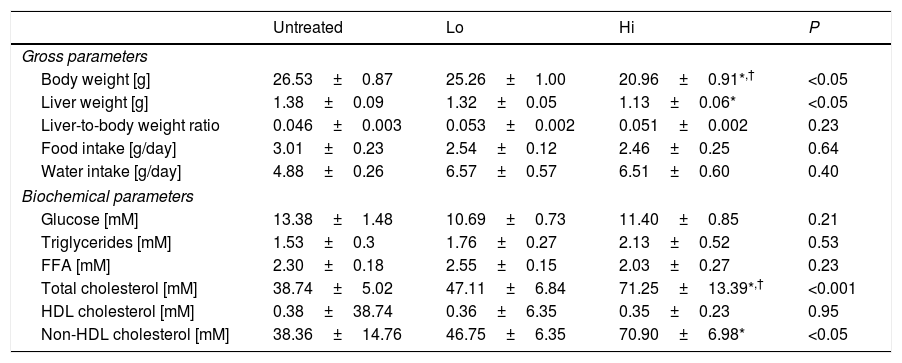
ResultsEffect of NAM on plasma lipids and lipoproteinsThe impact of administration of vitamin B3 derivatives in gross parameters of KOE mice is shown in Table 1. Palatability of water supplemented with vitamin treatments was well tolerated as revealed by the observed similar water intakes among the different groups of mice. NAM administration to mice did affect body or liver weight of NAM-treated mice in a dose-dependent manner (Table 1). Body weight gain shown by high-dose, NAM-treated mice was lower that in age-matched untreated mice (∼−5.6g, P<0.05), whereas no changes were observed in mice treated with the lower dose of NAM. Notably, the impact of NAM on body weight was not related to their daily food consumption as it did not differ with that shown by the untreated mice (Table 1). Although absolute liver weight of mice treated with high-dose of NAM was decreased compared with untreated mice, the liver weight relative to body weight ratio did not differ among groups.
Effect of NAM administration in KOE mice.
| Untreated | Lo | Hi | P | |
|---|---|---|---|---|
| Gross parameters | ||||
| Body weight [g] | 26.53±0.87 | 25.26±1.00 | 20.96±0.91*,† | <0.05 |
| Liver weight [g] | 1.38±0.09 | 1.32±0.05 | 1.13±0.06* | <0.05 |
| Liver-to-body weight ratio | 0.046±0.003 | 0.053±0.002 | 0.051±0.002 | 0.23 |
| Food intake [g/day] | 3.01±0.23 | 2.54±0.12 | 2.46±0.25 | 0.64 |
| Water intake [g/day] | 4.88±0.26 | 6.57±0.57 | 6.51±0.60 | 0.40 |
| Biochemical parameters | ||||
| Glucose [mM] | 13.38±1.48 | 10.69±0.73 | 11.40±0.85 | 0.21 |
| Triglycerides [mM] | 1.53±0.3 | 1.76±0.27 | 2.13±0.52 | 0.53 |
| FFA [mM] | 2.30±0.18 | 2.55±0.15 | 2.03±0.27 | 0.23 |
| Total cholesterol [mM] | 38.74±5.02 | 47.11±6.84 | 71.25±13.39*,† | <0.001 |
| HDL cholesterol [mM] | 0.38±38.74 | 0.36±6.35 | 0.35±0.23 | 0.95 |
| Non-HDL cholesterol [mM] | 38.36±14.76 | 46.75±6.35 | 70.90±6.98* | <0.05 |
Results are expressed as the means±SEM (n=5 mice per group). All analyses were made at 12 weeks of age. At the age of 8 weeks, the mice were challenged to high-fat, high cholesterol diet for 4 weeks. Food intake was measured at the end of the study as described in the Materials and methods section. Fasting plasma levels of the HDL fractions were determined in the plasma supernatants after precipitating with phosphotungstic acid (Roche); the non-HDL fraction was calculated by subtracting the HDL moiety to the total plasma. Differences between the mean values were assessed by the nonparametric a Kruskal–Wallis followed by Dunn's post-test; differences were considered significant when P<0.05. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice; FFA, free fatty acids; HDL, high-density lipoprotein; nd, not detectable.
Feeding mice with the high dose of NAM in water significantly raised the fasting plasma total cholesterol concentrations of treated mice compared to untreated mice (Table 1). This was due to an increase in the plasma concentration of non-HDL cholesterol. No differences were observed in total triglycerides among groups.
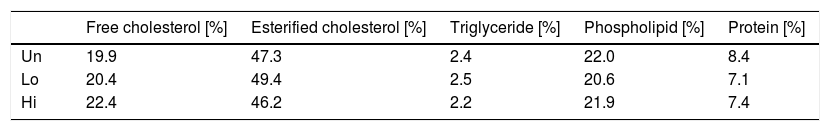
Composition analysis of non-HDL isolated by sequential ultracentrifugation revealed that NAM-related elevations in non-HDL were not due to changes in the relative chemical composition of this class of lipoproteins (Table 2).
Effect of NAM on the relative composition of non-HDL lipoproteins.
| Free cholesterol [%] | Esterified cholesterol [%] | Triglyceride [%] | Phospholipid [%] | Protein [%] | |
|---|---|---|---|---|---|
| Un | 19.9 | 47.3 | 2.4 | 22.0 | 8.4 |
| Lo | 20.4 | 49.4 | 2.5 | 20.6 | 7.1 |
| Hi | 22.4 | 46.2 | 2.2 | 21.9 | 7.4 |
Values shown are relative values for each components of non-HDL isolated by ultracentrifugation from fasting plasma of NAM-treated and untreated mice. A single pool composed of three mice per condition was used. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice.
Kinetic analysis using radiolabeled [3H]-cholesteryl oleoyl ether non-HDL indicated that NAM intake (highest dose) significantly lowered the fractional catabolic rate of intravenously injected [3H]-non-HDL (Fig. 1, panel A). However, the recovery of non-HDL-derived [3H]-cholesteryl oleoyl ether, expressed as percentage of the injected dose, only tended to be lower (P=0.15) in the livers of high-dose, NAM-treated than that measured in untreated mice (Fig. 1, panel B). No differences were observed in non-HDL synthesis rate between groups (Fig. 1, panel A, inset table).
![Effect of NAM on metabolic fate of non-HDL in plasma in KOE mice. In vivo kinetics of [3H]-cholesterol oleoyl ether non-HDL in plasma (panel A) and liver (panel B). Autologous 3H]-cholesterol oleoyl ether non-HDL were injected intravenously into fasted mice. The amounts of radioactivity remaining in the plasma (expressed as the average percentage±SEM, n=5–6 mice) were indicated at the indicated times after injection over a period of 24h. Inset: FCR and synthetic rates of the different mouse groups. Oral fat gavage (OFG) assays in NAM-treated and untreated mice. An olive oil-based emulsion containing radiolabeled [3H]-cholesterol was prepared and oral gavaged into KOE mice. Radiolabeled [3H]-cholesterol was, respectively, measured in plasma (panel C) and in the liver (panel D) after a single bolus of 200μL of radiolabeled olive oil-based emulsion (20μCi per mouse) at the times indicated over a period of 24h. Results are expressed as the average percentage vs. injected dose±SEM of individual animals (n=5 mice at each time point). In panels A and B, differences between mean values were assessed by the nonparametric U Mann–Whitney test. Specifically, *P<0.05 compared with the untreated group. Inset: area under the curve of [3H]-cholesterol non-HDL after the OFG of the different mouse groups. Each data represents the mean±SEM of 5 mice. In panels C and D, differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn](https://static.elsevier.es/multimedia/02149168/0000003100000006/v3_202004080610/S0214916819300476/v3_202004080610/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UiiaIbRDhkSNa5s/h9lbNFf+NuO/Bm/5xESvPPTQH5ELOU6r25bFQn+0CTYAKQ8xjJnGa6yheCmIhETj+82OBbaGot5SSsvaQkxbtOidiEFHBHjACYXJcJ+vcDQURstBBbjlHGCipcBYdftraZ11ZPBCEkNS0UlPjPM6FA1XQaj2hIKimladuYn/3wG0Evc6KTBI16LGpU+s0JdBFa7m3bAvWOuxFB2d6wjDztg+3ESH "Effect of NAM on metabolic fate of non-HDL in plasma in KOE mice. In vivo kinetics of [3H]-cholesterol oleoyl ether non-HDL in plasma (panel A) and liver (panel B). Autologous 3H]-cholesterol oleoyl ether non-HDL were injected intravenously into fasted mice. The amounts of radioactivity remaining in the plasma (expressed as the average percentage±SEM, n=5–6 mice) were indicated at the indicated times after injection over a period of 24h. Inset: FCR and synthetic rates of the different mouse groups. Oral fat gavage (OFG) assays in NAM-treated and untreated mice. An olive oil-based emulsion containing radiolabeled [3H]-cholesterol was prepared and oral gavaged into KOE mice. Radiolabeled [3H]-cholesterol was, respectively, measured in plasma (panel C) and in the liver (panel D) after a single bolus of 200μL of radiolabeled olive oil-based emulsion (20μCi per mouse) at the times indicated over a period of 24h. Results are expressed as the average percentage vs. injected dose±SEM of individual animals (n=5 mice at each time point). In panels A and B, differences between mean values were assessed by the nonparametric U Mann–Whitney test. Specifically, *P<0.05 compared with the untreated group. Inset: area under the curve of [3H]-cholesterol non-HDL after the OFG of the different mouse groups. Each data represents the mean±SEM of 5 mice. In panels C and D, differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn")
Effect of NAM on metabolic fate of non-HDL in plasma in KOE mice. In vivo kinetics of [3H]-cholesterol oleoyl ether non-HDL in plasma (panel A) and liver (panel B). Autologous 3H]-cholesterol oleoyl ether non-HDL were injected intravenously into fasted mice. The amounts of radioactivity remaining in the plasma (expressed as the average percentage±SEM, n=5–6 mice) were indicated at the indicated times after injection over a period of 24h. Inset: FCR and synthetic rates of the different mouse groups. Oral fat gavage (OFG) assays in NAM-treated and untreated mice. An olive oil-based emulsion containing radiolabeled [3H]-cholesterol was prepared and oral gavaged into KOE mice. Radiolabeled [3H]-cholesterol was, respectively, measured in plasma (panel C) and in the liver (panel D) after a single bolus of 200μL of radiolabeled olive oil-based emulsion (20μCi per mouse) at the times indicated over a period of 24h. Results are expressed as the average percentage vs. injected dose±SEM of individual animals (n=5 mice at each time point). In panels A and B, differences between mean values were assessed by the nonparametric U Mann–Whitney test. Specifically, *P<0.05 compared with the untreated group. Inset: area under the curve of [3H]-cholesterol non-HDL after the OFG of the different mouse groups. Each data represents the mean±SEM of 5 mice. In panels C and D, differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn's post-test; differences were considered significant when P<0.05. Specifically, *P<0.05 vs. untreated group. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice.
Similar results were obtained in an independent experiment that evaluated the fate of oral gavaged [3H]-cholesterol over a period of 24h (Fig. 1, panel C). In this case, the relative content of [3H]-cholesterol was found significantly reduced in the livers collected over a 24-h period from high-dose, NAM-treated mice (Fig. 1, panel D).
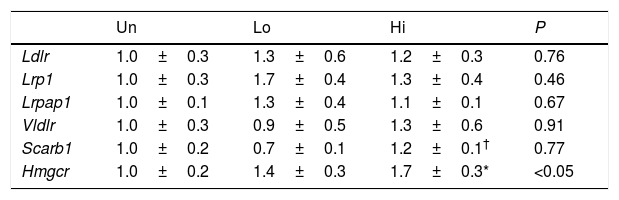
Impaired clearance of plasma non-HDL observed in NAM-treated mice was not accompanied by changes in the mRNA levels of key molecular targets involved in lipoprotein uptake of this class of lipoproteins (Table 3). However, the gene expression of Hmgcr was up-regulated in the livers of mice treated with the highest dose of NAM (1.7-fold, P<0.05).
Hepatic gene expression profile of molecular targets involved in non-HDL clearance and cholesterol synthesis.
| Un | Lo | Hi | P | |
|---|---|---|---|---|
| Ldlr | 1.0±0.3 | 1.3±0.6 | 1.2±0.3 | 0.76 |
| Lrp1 | 1.0±0.3 | 1.7±0.4 | 1.3±0.4 | 0.46 |
| Lrpap1 | 1.0±0.1 | 1.3±0.4 | 1.1±0.1 | 0.67 |
| Vldlr | 1.0±0.3 | 0.9±0.5 | 1.3±0.6 | 0.91 |
| Scarb1 | 1.0±0.2 | 0.7±0.1 | 1.2±0.1† | 0.77 |
| Hmgcr | 1.0±0.2 | 1.4±0.3 | 1.7±0.3* | <0.05 |
Results are expressed as the means±SEM (n=5 mice per group). The signal of untreated (Un) mice was set at a normalized value of 1 arbitrary unit. Differences between the mean values were assessed by the nonparametric a Kruskal–Wallis followed by Dunn's post-test; differences were considered significant when P<0.05. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice.
Cholesterol efflux ex vivo to either whole plasma or HDL obtained by precipitation with phosphotungstic acid was not changed by the administration of NAM (Fig. 2, panel A). To ascertain the impact of NAM on m-RCT, radiolabeled cholesterol was loaded in macrophages and injected into mice. The administration of NAM at the highest dose exhibited higher relative levels of plasma [3H]-cholesterol compared with untreated mice (Fig. 2, panel B), with counts primarily associated with the non-HDL fraction. Consistent with the view of previous kinetic studies, high-dose, NAM-treated mice exhibited lower relative hepatic levels of [3H]-cholesterol 48h after the injection of radiolabeled macrophages (Fig. 2, panel C). [3H]-tracer counts (both in the form of cholesterol and bile acids) in feces of high-dose, NAM-treated mice were also lower than those of untreated or low-dose, NAM-treated mice (Fig. 2, panel D).
![Effect of NAM over m-RCT in KOE mice. Panel A: cholesterol efflux ability mediated by total plasma and plasma HDL. Mouse plasma or HDL from ApoB-depleted plasma were added to the culture medium of J774A.1 cells loaded with [3H]-cholesterol. The percentage of cholesterol efflux was determined. Panels B–D: macrophage-to-plasma m-RCT remained unchanged in NAM-treated mice, whereas liver-to-feces m-RCT was decreased in NAM-treated mice. Individually housed mice were injected i.p. with [3H]-cholesterol-labeled J774A.1 mouse macrophages, and the distribution of counts into different compartments was determined 48h after the injection. In all panels, results are the mean±standard error of 5–6 mice and are expressed as % of injected dose, taking as a reference the % vs. injected dose determined in untreated (Un) mice (vs. Un). Panel B: plasma levels of [3H]-cholesterol in total plasma (Un: 11.48±0.96% vs. injected dose) and in plasma HDL (Un: 0.18±0.03% vs. injected dose). Panel C: hepatic levels of [3H]-cholesterol (Un: 14.08±1.73% vs. injected dose). Panel D: fecal total [3H]-tracer (Un: 1.19±0.24% vs. injected dose). Differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn](https://static.elsevier.es/multimedia/02149168/0000003100000006/v3_202004080610/S0214916819300476/v3_202004080610/en/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcxT+B4FjbiPdoCJto3d5S/Ch6vvG+Cp80/Jya7nfY0UiiaIbRDhkSNa5s/h9lbNFf+NuO/Bm/5xESvPPTQH5ELOU6r25bFQn+0CTYAKQ8xjJnGa6yheCmIhETj+82OBbaGot5SSsvaQkxbtOidiEFHBHjACYXJcJ+vcDQURstBBbjlHGCipcBYdftraZ11ZPBCEkNS0UlPjPM6FA1XQaj2hIKimladuYn/3wG0Evc6KTBI16LGpU+s0JdBFa7m3bAvWOuxFB2d6wjDztg+3ESH "Effect of NAM over m-RCT in KOE mice. Panel A: cholesterol efflux ability mediated by total plasma and plasma HDL. Mouse plasma or HDL from ApoB-depleted plasma were added to the culture medium of J774A.1 cells loaded with [3H]-cholesterol. The percentage of cholesterol efflux was determined. Panels B–D: macrophage-to-plasma m-RCT remained unchanged in NAM-treated mice, whereas liver-to-feces m-RCT was decreased in NAM-treated mice. Individually housed mice were injected i.p. with [3H]-cholesterol-labeled J774A.1 mouse macrophages, and the distribution of counts into different compartments was determined 48h after the injection. In all panels, results are the mean±standard error of 5–6 mice and are expressed as % of injected dose, taking as a reference the % vs. injected dose determined in untreated (Un) mice (vs. Un). Panel B: plasma levels of [3H]-cholesterol in total plasma (Un: 11.48±0.96% vs. injected dose) and in plasma HDL (Un: 0.18±0.03% vs. injected dose). Panel C: hepatic levels of [3H]-cholesterol (Un: 14.08±1.73% vs. injected dose). Panel D: fecal total [3H]-tracer (Un: 1.19±0.24% vs. injected dose). Differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn")
Effect of NAM over m-RCT in KOE mice. Panel A: cholesterol efflux ability mediated by total plasma and plasma HDL. Mouse plasma or HDL from ApoB-depleted plasma were added to the culture medium of J774A.1 cells loaded with [3H]-cholesterol. The percentage of cholesterol efflux was determined. Panels B–D: macrophage-to-plasma m-RCT remained unchanged in NAM-treated mice, whereas liver-to-feces m-RCT was decreased in NAM-treated mice. Individually housed mice were injected i.p. with [3H]-cholesterol-labeled J774A.1 mouse macrophages, and the distribution of counts into different compartments was determined 48h after the injection. In all panels, results are the mean±standard error of 5–6 mice and are expressed as % of injected dose, taking as a reference the % vs. injected dose determined in untreated (Un) mice (vs. Un). Panel B: plasma levels of [3H]-cholesterol in total plasma (Un: 11.48±0.96% vs. injected dose) and in plasma HDL (Un: 0.18±0.03% vs. injected dose). Panel C: hepatic levels of [3H]-cholesterol (Un: 14.08±1.73% vs. injected dose). Panel D: fecal total [3H]-tracer (Un: 1.19±0.24% vs. injected dose). Differences between mean values were assessed by the nonparametric Kruskal–Wallis test followed by Dunn's post-test; differences were considered significant when P<0.05. Specifically, *P<0.05 vs. untreated group; or †P<0.05 vs. low-dose, NAM-treated mice. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice.
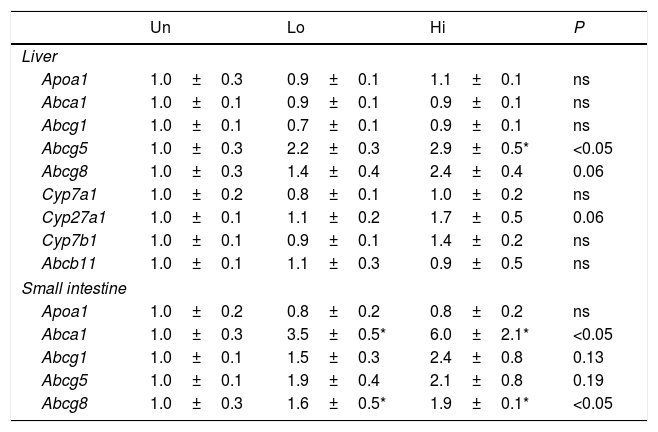
The gene expression of several transporters involved in cholesterol and bile acid homeostasis is shown in Table 4. Several members of the ABC transporters family (i.e., Abca1, Abcg1, Abcg5, and Abcg8) as well as other targets involved in bile acid synthesis (i.e., Cyp27a1) were analyzed. Abcg5 was up-regulated in the livers of high-dose, NAM-treated mice, but not in the small intestine, where it just showed a trend to be elevated. On the other hand, Abcg8 was up-regulated in the small intestine, whereas it just shows a trend to be elevated in the liver of high-dose, NAM-treated mice. The hepatic expression of genes involved in bile acid synthesis (Cyp7a1, Cyp27a1) and hepatobiliary transport (Abcb11) did not differ among groups. The gene expression of Abcg1 and Apoa1 in either the liver or small intestine was not influenced by NAM. Notably, the gene expression of Abca1 was highly up-regulated in the small intestine of NAM-treated mice.
Hepatic and intestinal gene expression profile of molecular targets involved in m-RCT.
| Un | Lo | Hi | P | |
|---|---|---|---|---|
| Liver | ||||
| Apoa1 | 1.0±0.3 | 0.9±0.1 | 1.1±0.1 | ns |
| Abca1 | 1.0±0.1 | 0.9±0.1 | 0.9±0.1 | ns |
| Abcg1 | 1.0±0.1 | 0.7±0.1 | 0.9±0.1 | ns |
| Abcg5 | 1.0±0.3 | 2.2±0.3 | 2.9±0.5* | <0.05 |
| Abcg8 | 1.0±0.3 | 1.4±0.4 | 2.4±0.4 | 0.06 |
| Cyp7a1 | 1.0±0.2 | 0.8±0.1 | 1.0±0.2 | ns |
| Cyp27a1 | 1.0±0.1 | 1.1±0.2 | 1.7±0.5 | 0.06 |
| Cyp7b1 | 1.0±0.1 | 0.9±0.1 | 1.4±0.2 | ns |
| Abcb11 | 1.0±0.1 | 1.1±0.3 | 0.9±0.5 | ns |
| Small intestine | ||||
| Apoa1 | 1.0±0.2 | 0.8±0.2 | 0.8±0.2 | ns |
| Abca1 | 1.0±0.3 | 3.5±0.5* | 6.0±2.1* | <0.05 |
| Abcg1 | 1.0±0.1 | 1.5±0.3 | 2.4±0.8 | 0.13 |
| Abcg5 | 1.0±0.1 | 1.9±0.4 | 2.1±0.8 | 0.19 |
| Abcg8 | 1.0±0.3 | 1.6±0.5* | 1.9±0.1* | <0.05 |
Results are expressed as the means±SEM (n=5 mice per group). The signal of untreated (Un) mice was set at a normalized value of 1 arbitrary unit. Differences between the mean values were assessed by the nonparametric a Kruskal–Wallis followed by Dunn's post-test; differences were considered significant when P<0.05. Abbreviations used: Un, untreated mice; Lo, low-dose, NAM-treated mice; Hi, high-dose, NAM-treated mice.
There is no direct experimental evidence on the potential of NAM in contributing to m-RCT in vivo. This study reveals, for the first time to our knowledge, that NAM failed to improve m-RCT in KOE mice, with the underlying mechanisms possibly attributed to a defective uptake of non-HDL lipoproteins by the liver of treated mice.
The treatment of mice with the highest dose of NAM produced a significant reduction in the body weight. Supplementation of other NAD+ intermediates (i.e., nicotinamide riboside and nicotinamide mononucleotide) has shown favorable effects on health, protecting against high-fat induced obesity and increasing energy expenditure in vivo.24–26 However, our data contrast with a previous recent report showing that NAM treatment did not significantly alter body weight in mice.27 We speculate that the differences between this study and our findings might be explained, at least in part, by differences in the amount of NAM given to mice. Further research is warranted to elucidate the impact of NAM on the body weight in KOE mice.
Administration of NAM produced total cholesterol elevations mainly attributed to an increase in the plasma fraction of non-HDL. The direct analysis of the chemical composition of isolated non-HDL lipoproteins did not reveal quantitative changes among groups, thus suggesting that the observed differences might be rather due to an increased number of these lipoprotein particles in circulation. Supporting to this, kinetic analysis clearly showed a significant retention of non-HDL lipoproteins in plasma and defective hepatic uptake in NAM-treated mice. This finding was not explained by either functional liver defects (data not shown) or concomitant alterations in the gene expression of different molecular targets involved in the uptake of non-HDL lipoproteins, potentially suggesting the participation of posttranscriptional NAM-mediated mechanisms.
In our experimental setting, total and HDL-mediated cholesterol efflux ability, which is the first step of the m-RCT, was not influenced by NAM. Of note, we found a higher amount of macrophage-derived cholesterol in the non-HDL fraction, which could be transferred from HDL by the action of phospholipid transfer protein.28 Alternatively, LDL may also induce macrophage cholesterol efflux.29 Our data suggest that the relative accumulation of radiolabeled cholesterol in plasma, which was mainly attributed to a reduced clearance of circulating non-HDL lipoproteins, could be hampering the accumulation of hepatic pools of radiotracer to be eventually removed into feces over time. Supporting to this, our data showed reduced relative hepatic content of [3H]-cholesterol in high-dose, NAM-treated mice compared with either low-dose or untreated mice. Interestingly, this phenotype was accompanied by a concomitant up-regulation of Hmgcr in high-dose, NAM-treated mice. Cholesterol depletion leads to the transcriptional activation of target genes, which include Hmgcr.30 Potentially, the rise in the expression levels of Hmcgr could be partly attributed to decreased cellular cholesterol in the livers of mice treated with the highest dose of NAM.
The treatment of KOE mice with NAM rather worsened overall m-RCT process in vivo, despite showing, a significant up-regulation, or trend to be higher, in some of the liver X receptor (LXR) gene targets involved in the hepatobiliary trafficking of cholesterol. This finding suggested that LXR might be induced in tissues from NAM-treated mice. The mechanisms leading to such LXR induction were not studied in the present work, but might potentially involve sirtuin (Sirt)-1. First, NAM is a NAD+ precursor in vivo.31–33 Second, beneficial effects of other NAD+ intermediates (i.e., nicotinamide riboside and nicotinamide mononucleotide) have been attributed to sirtuin activation.25,26,34 Next, increased cellular levels of NAD preserve Sirt1 action35 and mimic the metabolic phenotype of mice with Sirt1 gain-of-function.36–38 In this regard, cholesterol elevations in plasma were also observed in Sirt1-overexpressing, Ldlr-deficient mice placed on a high-fat, high cholesterol diet.39 Lastly, Sirt1 positively regulates LXR.40 Supporting this notion, the administration of NAM to our KOE mice resulted in a significant increase in the hepatic content of NAD+ (K.A. Méndez-Lara, unpublished results). However, further research is still needed to demonstrate the contribution of NAM-mediated increase in NAD+ levels on the Sirt1 signaling in KOE mice.
Our study shows that NAM failed in driving the hepatobiliary component of m-RCT, at least in part, due to a decreased hepatic uptake of circulating non-HDL. These findings may cast doubts on the potential favorable use of NAM in therapies addressed to treat atherosclerosis.
FundingThis work was funded by a grant from Fundación Española de Arteriosclerosis para Nutrición “Manuel de Oya” 2015 (to J.J.) and by Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III (ISCIII), grants FIS PI17-00232 (to J.J.), PI17-01362 (to N.A.), PI11/01076 (to F.B.-V.), PI16/00139 (to J.C.E.-G.), and FEDER “Una manera de hacer Europa”; and by La Marató de TV3 2016 (201602.30.31) (to N.A. and J.J.). J.J. is recipient of a Miguel Servet Type 2 contract (CPII18/00004; ISCIII). K.A.M.-L. was recipient of an AGAUR grant FI-DGR2014 (Generalitat de Catalunya). CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM) is a project of Instituto de Salud Carlos III (CB07/08/0016). Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau is accredited by the Generalitat de Catalunya as Centre de Recerca de Catalunya (CERCA).
Conflicts of interestThe authors declare no conflict of interest.