





Las hipocolesterolemias primarias (o hipobetalipoproteinemias) constituyen un trastorno infrecuente del metabolismo de las lipoproteínas que pueden obedecer a una predisposición poligénica o a una enfermedad monogénica. Entre estas, es posible diferenciar entre formas sintomáticas y asintomáticas, en las que, en ausencia de causas secundarias, la sospecha clínica inicial son concentraciones plasmáticas de ApoB por debajo del percentil 5 de la distribución por edad y sexo. En esta nota clínica describimos del diagnóstico diferencial de un caso de hipocolesterolemia asintomática. Estudiamos los datos clínicos de la paciente índice, así como su perfil lipídico y el de los familiares junto con los datos clínicos de estos que son relevantes para realizar el diagnóstico diferencial. Se realizó un estudio genético como prueba diagnóstica. El diagnóstico diferencial realizado sugirió una hipobetalipoproteinemia heterocigota por variantes de pérdida de función en PCSK9. La prueba diagnóstica puso de manifiesto, en la paciente índice, la presencia de una variante de cambio de pauta de lectura en PCSK9, en heterocigosis, de origen materno. Las concentraciones plasmáticas de colesterol de LDL y PCSK9 de la paciente y los familiares, fueron compatibles con la segregación de dicha variante. En conclusión, la prueba diagnóstica realizada permitió confirmar el diagnóstico de sospecha en el caso estudiado de hipobetalipoproteinemia familiar asintomática a causa de una variante de pérdida de función en el gen PCSK9.
Primary hypocholesterolemia (or hypobetalipoproteinemia) is a rare disorder of lipoprotein metabolism that may be due to a polygenic predisposition or a monogenic disease. Among these, it is possible to differentiate between symptomatic and asymptomatic forms, in which, in the absence of secondary causes, the initial clinical suspicion is plasma ApoB levels below the 5th percentile of the distribution by age and sex. Here we describe the differential diagnosis of a case of asymptomatic hypocholesterolemia. We studied proband's clinical data, the lipid profile of the proband and her relatives and the clinical data of the family relevant to carry out the differential diagnosis. We performed a genetic study as the diagnostic test. The information obtained from the differential diagnosis suggested a heterozygous hypobetalipoproteinemia due to PCSK9 loss-of-function variants. The diagnostic test revealed, in the proband, the presence of a heterozygous PCSK9 frame-shift variant of a maternal origin. Plasma levels of LDL cholesterol and PCSK9 of the patient and her relatives were compatible with the segregation of the variant revealed. In conclusion, the diagnostic test performed confirmed the suspected diagnosis of the proband as asymptomatic familial hypobetalipoproteinemia due to a loss-of-function variant in the PCSK9 gene.
Las hipocolesterolemias primarias constituyen un trastorno infrecuente del metabolismo de las lipoproteínas que pueden obedecer a una predisposición poligénica o a una enfermedad monogénica1,2. Mostramos un caso de hipocolesterolemia que fue presentado en la XVIII Reunión Nacional de Hipertrigliceridemias y VIII Reunión de las Unidades de Lípidos de la SEA (Toledo), en formato de caso cerrado.
Caso clínico cerradoMujer de 33 años que derivan a la Unidad de Lípidos por hipocolesterolemia (colesterol de LDL<50mg/dl). Entre sus antecedentes personales, niega alergias, hábitos tóxicos o intervenciones quirúrgicas. Diagnosticada de asma en tratamiento con formoterol/budesónida a demanda. Entre sus antecedentes familiares destaca únicamente el padre con obesidad, diabetes, hipertensión y policitemia primaria, siendo tratado con hidroxiurea, atorvastatina 20mg, metformina 850mg/12h, enalapril 20, bisoprolol 10 y trifusal 1 comprimidos/día. En la anamnesis la paciente estaba asintomática, sin diarrea ni otros síntomas gastrointestinales. En la exploración, presentó un peso de 58kg, talla 1,51 m, perímetro de cintura 79cm, presión arterial 130/75mmHg, no se hallaron xantelasmas ni xantomas. Cuello con buenos pulsos carotídeos, sin soplos. Auscultación cardiaca y pulmonar sin anomalías. Abdomen al mismo nivel que tórax, blando y depresible, no se palpan megalias. Extremidades con pulsos conservados.
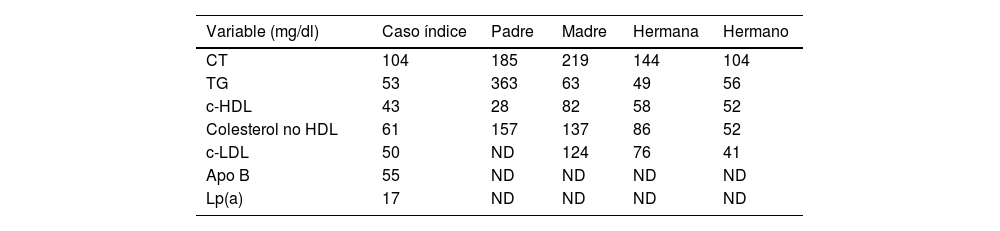
El perfil lipídico de la paciente y de sus familiares está recogido en la tabla 1; ni el caso índice ni sus familiares mostraron anomalías del perfil hepático, tiroideo o renal.
Analítica en ayunas del caso índice y familiares de primer grado
| Variable (mg/dl) | Caso índice | Padre | Madre | Hermana | Hermano |
|---|---|---|---|---|---|
| CT | 104 | 185 | 219 | 144 | 104 |
| TG | 53 | 363 | 63 | 49 | 56 |
| c-HDL | 43 | 28 | 82 | 58 | 52 |
| Colesterol no HDL | 61 | 157 | 137 | 86 | 52 |
| c-LDL | 50 | ND | 124 | 76 | 41 |
| Apo B | 55 | ND | ND | ND | ND |
| Lp(a) | 17 | ND | ND | ND | ND |
c-HDL: colesterol de HDL; c-LDL: colesterol de LDL; CT: colesterol total; ND: no determinado; TG: triglicéridos.
Se le realizó una ecografía abdominal que fue informada como hígado de tamaño normal con ecogenicidad homogénea sin datos indicativos de esteatosis hepática. Resto de la exploración sin anomalías.
Se procedió a una prueba diagnóstica.
Diagnóstico diferencialCon la información que disponemos, parece que estamos ante un caso de hipobetalipoproteinemia, que se define por la presencia de niveles plasmáticos de ApoB por debajo del percentil 5 para la edad y sexo3. Estas alteraciones analíticas pueden ser secundarias o primarias. Entre las secundarias se pueden destacar dietas vegetarianas muy estrictas, alcoholismo crónico o diversas enfermedades, como la malabsorción intestinal, enfermedades hepáticas, malnutrición o hipertiroidismo, así como secundario a tratamiento hipolipidemiante4. Ninguna de estas situaciones está presente en nuestro caso, por lo que parece razonable descartarlas.
Entre las causas primarias monogénicas destacan la abetaliproteinemia, la hipobetalipoproteinemia familiar, la enfermedad por retención de quilomicrones, la hipolipidemia familiar combinada y la hipobetalipoproteinemia por pérdida de función de PCSK92. Dentro de ellas, haremos una distinción entre aquellas hipobetalipoproteínas primarias sintomáticas y asintomáticas.
Entre las sintomáticas, encontramos la abetalipoproteinemia, una hipolipidemia autosómica recesiva (AR) producida por variantes del gen MTTP con niveles de lípidos en plasma muy bajos, casi ausentes y déficit de vitaminas liposolubles. Se caracterizada por la presencia desde el periodo neonatal de un síndrome de malabsorción de grasas, con esteatorrea, vómitos, retraso en el crecimiento, así como retinitis, ataxia grave, pueden desarrollar enfermedad hepática y es característica la presencia de acantocitosis. La enfermedad por retención de quilomicrones de herencia AR producida por mutaciones en el gen SAR1B y la hipobetalipoproteinemia familiar homocigota o heterocigota compuesta, de herencia autosómica dominante (AD) por mutación del gen APOB, son 2 enfermedades clínica y bioquímicamente prácticamente indistinguibles de la abetalipoproteinemia —salvo por los niveles de triglicéridos (TG) normales que presenta la enfermedad por retención de quilomicrones. Por tanto, el diagnóstico de estas hipobetalipoproteinemias requeriría un estudio genético1,5. En cualquier caso, estas 3enfermedades las podemos descartar de forma razonable en el caso índice dado que la paciente se encuentra asintomática.
Dentro de las hipobetalipoproteinemias asintomáticas, está la hipobetaliproteinemia familiar heterocigota4, de herencia AD por variantes patogénicas en el gen APOB que originan proteínas truncadas. Los pacientes portadores son generalmente asintomáticos, pero pueden desarrollar enfermedad hepática de naturaleza esteatósica con hipertransaminasemia, c-LDL bajo, pero no indectectable, habitualmente por debajo de 89mg/dl, con concentraciones de TG normales. Cuando la variante de APOB tiene menor longitud que la apoB48 se ve comprometida la absorción de grasa6. También es asintomática la hipolipidemia familiar combinada7, de herencia AD por variantes en el gen ANGPTL3. Los casos homocigotos presentan concentraciones de c-LDL, c-HDL y TG muy bajos, y los heterocigotos presentan una reducción de aproximadamente el 50% de las cifras de CT, c-LDL y TG, con c-HDL relativamente normal. Finalmente, entre las formas asintomáticas también está la hipobetaliproteinemia familiar causada por variantes de pérdida de función en PCSK98, de herencia autosómica dominante, con concentraciones de c-LDL baja pero detectable (21-40% inferiores a la normal). Estas 3hipolimidemias asintomáticas parecen asociarse con disminución del riesgo cardiovascular.
Según lo expuesto, ante el caso que se nos presenta, descartaríamos hipolipidemia familiar combinada dado que no presentan concentraciones muy bajas de TG. Además, la forma homocigota tendría concentraciones de CT y c-LDL muchos más bajas. Respecto a la hipobetalipoproteinemia familiar heterocigota, a pesar de que los pacientes suelen estar asintomáticos, también pueden presentar enfermedad hepática, que parece descartada en la paciente y en los familiares. Por tanto, nuestro diagnóstico de sospecha es la hipobetalipoproteinemia heterocigota por variantes de pérdida de función en PCSK9.
Resolución del casoLa prueba diagnóstica fue la secuenciación masiva paralela (Genológica, Málaga, España) que mostró la presencia de una variante en el exón 9 de PCSK9, de cambio de pauta de lectura: c.1378delG; p.Val460Tyrfs (rs749090549), no descrita previamente en la literatura y con una frecuencia alélica<0,001. Según los criterios de la Colegio Americano de Medicina Genética y Genómica (ACMG) esta variante está clasificada como variante de significado incierto al tener un único criterio de patogenicidad, por el cambio de marco de lectura producido, y haberse encontrado en sujetos sanos (https://varsome.com/variant/hg38/rs749090549?annotation-mode=germline). Para comprobar si esta variante segregaba en la familia, se tomaron muestras del caso índice, de los padres y un hermano, y fueron analizadas por la técnica de análisis de curvas de fusión de alta resolución (High Resolution Melting o HMR)9, en el laboratorio de Lípidos y Arteriosclerosis (CIMES, Universidad de Málaga). Esta metodología permitió identificar la presencia de la variante en la madre y en el hermano analizado, además de confirmarla en el caso índice (fig. 1). La concentración de la proteína PCSK9 circulante se midió mediante un ensayo por inmunoabsorción ligado a enzimas (Human Proprotein Convertase 9/PCSK9 Quantikine ELISA Kit [DPC900], R&D Systems®, Inc., Minneapolis, Estados Unidos). La densidad óptica fue leída a una longitud de onda de 450nm utilizando el lector de microplacas SPECTROstar Nano (BMG Labtech, gmbh, Offemburg, Alemania). Las concentraciones de las muestras se determinaron por interpolación a partir de la curva estándar generada con las muestras patrón suministradas por el fabricante con una dilución 1:20 de las muestras de suero (Laboratorio BiosferTeslab, Reus, Tarragona, España). Los resultados muestran que los portadores de la variante identificada tenían concentraciones más bajas de lo habitual, dado que, con el ensayo utilizado, los valores de normalidad son 313±71,5 ng/ml.
 en LDL (mg/dl) y de PCSK9 (ng/ml).")
En relación con el caso que nos ocupa son pertinentes los siguientes comentarios. La hipobetalipoproteinemia familiar es infrecuente pero no puede ser considerada una enfermedad rara. Datos recientes del Biobanco del Reino Unido y del NHBLI americano, sobre un total de 209.537 muestras, indican que el 0,4% de las mismas corresponden a pacientes con variantes que producen proteínas truncadas en apolipoproteína B o en PCSK9, siendo esta última más frecuente que la primera10. Finalmente, en términos de riesgo vascular, la presencia de hipobetalipoprotenemia familiar reduce el riesgo de forma notable, entre un 80 y un 50%8,10, por lo que las estrategias de prevención deben mantenerse en estos pacientes.
En resumen, presentamos un caso de hipobetalipoproteinemia familiar asintomática debido a una variante de pérdida de función en el gen PCSK9.
FinanciaciónFondos propios del grupo (CTS-159) provenientes del Plan Andaluz de Investigación, Desarrollo e Innovación (PAIDI).
La Universidad de Málaga/CBUA ha financiado la publicación en abierto de este artículo.
Conflicto de interesesLos autores declaran no tener conflictos de interés aplicables a esta publicación.
Al caso índice y sus familiares estudiados, que dieron su consentimiento para la publicación anónima.