








La homocisteína (HCY) es un aminoácido cuya elevación sanguínea se asocia con el desarrollo de enfermedades de tipo vascular, neurológico y reproductivo. Sus niveles plasmáticos varían en función de la raza, el sexo, la edad y otros factores ambientales. En la presente revisión se aborda su metabolismo, su fisiopatología y las consecuencias clínicas de su elevación. Un énfasis especial se presta al empleo del ratón como modelo experimental en este campo, ya que su uso ha puesto de manifiesto la importancia de la dieta en la regulación de la HCY. Esto, unido al desarrollo de animales modificados genéticamente con hiperhomocisteinemia, está permitiendo una rápida caracterización de los mecanismos moleculares implicados en la acción in vivo de la HCY. Además, la combinación de estos modelos con otros modificados genéticamente permite definir la influencia de la combinación de factores de riesgo en el desarrollo de diversas patologías. A su vez, la exploración en estos nuevos modelos de factores ambientales y/o farmacológicos contribuye de este modo a explicar muchas de las evidencias epidemiológicas en humanos así como el tratamiento más adecuado para cada condición.
Homocysteine is an amino acid whose plasma levels are associated with the development of vascular, neurologic and reproductive diseases. Plasma levels show a wide range of values according to age, sex, race and other environmental factors. The present review discusses homocysteine metabolism and physiopathology and the clinical consequences of increased levels of this substance.
Special emphasis has been placed on the use of mice as an experimental animal model in this field, since their use has unveiled the contribution of dietary modifications on plasma homocysteine levels. These findings, together with the generation of genetically-modified mice as models of hyperhomocysteinemia, are allowing rapid progress to be made in the characterization of the in vivo molecular mechanisms of homocysteine action. Crosses among these genetically-modified mice and others with different deleted genes will increase knowledge of the influence of the combination of several risk factors on pathological development. In these models, research into new environmental or pharmacological factors may yield results that could explain epidemiological findings in humans and help in the design of new treatments for specific clinical settings.
En los últimos años ha crecido el interés por la hiperhomocisteinemia (HHCY) como factor de riesgo asociado a las enfermedades cardiovasculares1–4 en un intento por conocer mejor los mecanismos etiopatogénicos de estas y dado que su morbimortalidad continúa siendo particularmente importante en los países industrializados y, en concreto, en España5.
El tabaquismo, las dislipemias, la hipertensión arterial y la diabetes son factores de riesgo clásicos que se asocian con la aparición prematura de la enfermedad cardiovascular6; sin embargo, algunos pacientes con manifestaciones clínicas de aterosclerosis precoz no presentan ninguno de estos factores, con la excepción del hallazgo de una HHCY.
La hipótesis de que la homocisteína (HCY) estaba implicada en las lesiones vasculares fue propuesta por McCully7, al observar en pacientes con severa HHCY sin tratar un 50% más de posibilidades de sufrir un problema vascular antes de los 30 años, como ictus, infarto de miocardio o tromboembolismo venoso8. El riesgo se reducía cuando se empleaban tratamientos para disminuir la HCY, aunque no se alcanzasen los niveles normales9.
Metabolismo de la homocisteínaLa HCY es un aminoácido azufrado que se forma durante el catabolismo de la metionina. Las estructuras de ambos aminoácidos están reflejadas en la figura 1. Este último aminoácido esencial, aportado por las proteínas de la dieta, es un componente de péptidos y proteínas, y para su utilización metabólica se activa a S-adenosil metionina, coenzima que desempeña un papel importante en la transferencia de grupos metilos. La S-adenosil metionina transfiere su grupo metilo y se transforma en S-adenosil HCY, que posteriormente se hidroliza a HCY en las células. Una vez sintetizada, la HCY puede seguir varias rutas metabólicas dentro del organismo: la ruta de transulfuración, la ruta de remetilación y la excreción por la orina10,11. Un esquema del metabolismo de la HCY se recoge en la figura 2.


Metabolismo de la homocisteína a partir de la metionina proveniente del catabolismo proteico. BHMT: betaína homocisteína metiltransferasa; CBS: cistationina betasintasa; CGL: cistationina gammaliasa; MAT: metionina adenosil transferasa; Metil THF: metiltetrahidrofolato; Metilen THF: metilentetrahidrofolato; MS: metionina sintasa; MTHFR: metilentetrahidrofolato reductasa; SAH: S-adenosil homocisteína hidrolasa; SMMT: S-metilmetionina metiltransferasa; STHM: serina transhidroximetilasa; THF: tetrahidrofolato.
La HCY sigue el camino de transulfuración cuando existe un exceso de metionina o cuando se requiere la síntesis de cisteína. En esta vía, la HCY se condensa con una molécula de serina para formar cistationina en una reacción catalizada por la cistationina betasintasa (CBS), que utiliza la vitamina B6 como cofactor. A continuación, la cistationina gammaliasa (CGL) con la ayuda de nuevo de la vitamina B6 cataliza la hidrólisis de cistationina para dar lugar al alfacetobutirato y la cisteína12,13. Ambas enzimas (CBS y CGL) son responsables de la biosíntesis del sulfuro de hidrógeno (H2S) en mamíferos. La CBS puede generar la desulfuración de varias formas: al actuar sobre la cisteína para generar serina o por condensación de dos cisteínas para generar lantionina. La participación de la CBS en la biosíntesis del H2S disminuye en condiciones de HHCY14. La CBS también es un sensor de CO, y mediante la liberación de H2S participa en la regulación del metabolismo de la hemooxigenasa (HO)15. La CGL condensa dos moléculas de HCY para formar H2S y hololantionina. Este último proceso aumenta en la medida en que se eleva la HHCY y es particularmente relevante en concentraciones superiores a 200μmol/l16.
En la ruta de remetilación, la HCY se transforma en metionina. En el hombre, aproximadamente el 50% de la HCY sufre este destino en condiciones normales y resulta particularmente importante en situaciones de baja ingesta proteica17,18. En la mayoría de los tejidos, la remetilación se produce a través de una reacción catalizada por la metionina sintasa (MS), que requiere de vitamina B12 como cofactor y de metiltetrahidrofolato como cosustrato. Esta vía necesita de un adecuado aporte de ácido fólico y de la actividad de otra enzima denominada metilentetrahidrofolato reductasa (MTHFR)19,20. También se puede producir la remetilación a través de la betaína HCY metiltransferasa, reacción restringida al hígado, al riñón y a las glándulas suprarrenales y que, además, no requiere la presencia de vitaminas. Más recientemente se ha propuesto otra reacción de metilación en la cual el donante es la S-metil metionina y la enzima participante es la S-metil metionina HCY metiltransferasa21. Sin embargo, su significado biológico no ha sido establecido. La acción conjunta de estas reacciones de metilación permite conservar la metionina, con una implicación variable tal como se ha descrito previamente. De este modo se garantiza una adecuada concentración de la S-adenosil metionina, considerada como un donador universal de grupos metilo.
Homocisteína en el plasmaLa HCY se presenta en el plasma de tres formas diferentes. La forma mayoritaria, que representa el 70% del total, está unida a varias proteínas, especialmente a la albúmina, a la hemoglobina y a la alfaglobulina22. Se encuentra en forma de dímero unido por un puente disulfuro en un 25%, y en un 5% se encuentra en forma libre reducida o combinada con otros tioles23.
Cuando se habla de HCY plasmática total, se engloban las tres formas. Son varios los métodos para medir la HCY y están basados en distintas técnicas de cromatografía líquida24–26. Recientemente se ha incorporado un inmunoensayo de polarización fluorescente que permite la automatización de los ensayos27.
La conservación de las muestras sanguíneas es fundamental. En muestras almacenadas a temperatura ambiente durante más de 4h se observan incrementos de hasta un 35% en la concentración de la HCY. Se considera que dicho aumento es debido a la influencia del deterioro de los eritrocitos28. Para evitar este fenómeno, las muestras se conservan en hielo y se requiere la separación del plasma lo más rápidamente posible.
Los valores de referencia varían según el método utilizado, la edad y el sexo de la población evaluada, y oscilan entre 5–15μmol/l29,30. En este sentido, los valores de HCY son más elevados en el hombre que en la mujer y, a su vez, son mayores en las mujeres posmenopáusicas que en las premenopáusicas31. La diferencia de valores entre sexos podría deberse a un efecto hormonal o estar relacionada con la diferente masa muscular.
La HCY plasmática aumenta también con la edad32, fenómeno que podría estar originado por una disminución de los niveles de los cofactores enzimáticos, una disfunción renal y/o una disminución de la actividad de la CBS33,34. Igualmente, se han descrito diferencias étnicas en los niveles de HCY, siendo inferiores en la raza negra que en la raza blanca o la asiática35,36.
La HHCY se define normalmente a través de unos valores arbitrarios que normalmente son los que están por encima del percentil 95 o por encima de los valores medios obtenidos en individuos sanos en ayunas más dos veces la desviación estándar y considerados los valores de referencia37. Según los valores hallados, se clasificaría en:
- •
HHCY ligera: 16–30μmol/l.
- •
HHCY moderada: 31–100μmol/l.
- •
HHCY severa: >100μmol/l.
Existe una correlación entre los niveles de HHCY y el incremento del riesgo en la enfermedad coronaria38,39, el infarto de miocardio40,41, la enfermedad oclusiva periférica32, la enfermedad oclusiva cerebral42 y la enfermedad oclusiva vascular retinal43. En algunas poblaciones puede ser particularmente importante. Así, Selhub et al en el «Framingham Heart Study» encontraron que la incidencia de HHCY era de un 29,3% en un grupo de 1.041 individuos adultos de entre 67–96 años de edad32. También encontraron una correlación lineal entre el riesgo de infarto y los niveles de HCY. La HHCY se reveló, asimismo, como un factor de riesgo para la trombosis venosa, particularmente en las mujeres44. La presencia de niveles de HCY superiores a 12μmol/l está relacionada directamente con la progresión de la placa coronaria45. Sin embargo, se ha generado cierta controversia a raíz de los resultados obtenidos en estudios clínicos de tratamiento de la HHCY con suplementos de vitaminas del grupo B, donde se observó una disminución de la concentración plasmática de HCY pero no del riesgo vascular, lo cual vendría a enfatizar la mayor relación de la HHCY con el riesgo de enfermedad cerebrovascular que con el de enfermedad cardiovascular (una detallada revisión de estos últimos aspectos se puede encontrar en el reciente trabajo de Méndez-González et al46). Se ha observado disfunción vasomotora endotelial en vasos de pequeña resistencia, como las arterias mesentéricas o las cerebrales, con leves aumentos en la concentración de HCY: entre 10–20μmol/l47–50. En cambio, en ratón, para producir daños en los grandes vasos, como la aorta o la carótida, sería necesario alcanzar concentraciones plasmáticas por encima de 20μmol/l51,52.
En modelos experimentales con HHCY también se han observado alteraciones estructurales en la pared de los vasos: hipertrofia, remodelado, mecánica vascular alterada, aumento de la fragilidad de los vasos53–56, aumento de formación de la íntima57 y trombosis acelerada58. En este sentido, se ha observado que la presencia de HHCY potencia el desarrollo de aterosclerosis en animales susceptibles de desarrollarla, como los ratones carentes de apolipoproteína E (ApoE)59. Sin embargo, ninguno de los modelos animales estudiados ha desarrollado aterosclerosis espontáneamente. También se ha demostrado que una dieta con alta cantidad de metionina puede causar aterosclerosis en ausencia de aumento de HCY en el plasma60. Por todo lo anterior, se considera la homocisteinemia elevada como un factor de riesgo independiente para la enfermedad cardiovascular.
Homocisteína y enfermedad renalLa insuficiencia renal crónica es otra patología en la que se ha descrito la presencia de niveles elevados de HCY61. Estos autores encontraron una correlación positiva entre los niveles de HCY y de creatinina, lo cual podría ser debido a una disminución del metabolismo renal y/o de su excreción62. Igualmente, se observaron concentraciones de HCY particularmente elevadas (26,6±1,5μmol/l) en pacientes con enfermedad renal terminal sometidos a hemodiálisis34. El aporte de H2S redujo el daño renal inducido por la HHCY63, al igual que el estrés oxidativo. Esto sugiere que el aporte de NaHS/H2S puede reducir el estrés oxidativo de la HCY y ser una herramienta terapéutica64.
La homocisteína como factor de riesgo en reproducciónDiversos estudios epidemiológicos han mostrado una asociación entre alteraciones del metabolismo de la metionina y la HCY y casos de embarazos fallidos con aborto espontáneo, infartos de la placenta o bajo peso al nacer65,66. También se han descrito niveles altos de HCY en casos de gestaciones complicadas con enfermedades vasculares de la placenta o preeclampsia en comparación con los encontrados en embarazos normales67,68.
La enzima CBS se expresa en el hígado y en la decidua, mientras que la enzima MS se expresa en todos los tejidos embrionarios69,70. En el ratón carente de la enzima CBS, las hembras eran infértiles71. Para caracterizar el fallo reproductivo, nuestro grupo realizó el trasplante de ovarios de hembras carentes de CBS a hembras normales ovariectomizadas y se obtuvieron camadas normales en cuanto al número de crías, indicando que el problema que produce infertilidad era consecuencia de la falta de CBS y del aumento de HCY en el útero gestante72. Estos resultados plantean el reto de investigar si el útero humano es tan sensible al efecto de la HCY.
Homocisteína y enfermedades neurológicasLa elevación de la HCY en el plasma se ha relacionado de manera convincente con enfermedades geriátricas del cerebro, como declive de consciencia73–75, enfermedades cerebrovasculares76,77, demencia vascular78,79 y enfermedad de Alzheimer80–84. Además, podría estar relacionada con otras enfermedades como la depresión85 y la esquizofrenia86,87. En la mayoría de estos estudios, las asociaciones continúan siendo fuertes aunque se ajusten parámetros importantes en la HHCY, como la función renal y el estatus vitamínico88.
Se han propuesto cuatro posibles mecanismos de lesión neuronal debida a la HHCY; el primero, la posibilidad de una acción neurotóxica de la HCY mediante la alteración de los neurotransmisores o la producción de excitotoxicidad en las neuronas89–91. En concreto, se ha descrito una competencia del compuesto por los receptores de GABA-A y una activación de la ruta de la quinasa ERK92. El segundo mecanismo propone que el aumento de HCY indica una alteración metabólica que afecta al cerebro a través de la acumulación de S-adenosil HCY, un inhibidor de las reacciones de metilación vitales para las funciones neurológicas93. La tercera posibilidad es la relación de la HCY con enfermedades oclusivas de los vasos sanguíneos mediante el daño de la pared vascular o el impedimento de la coagulación sanguínea. La cuarta contempla una acción sobre la microglia94. En estudios in vivo no se ha observado una relación directa entre el aumento de HCY y las lesiones cerebrales, lo que se explicaría, en parte, por la baja concentración de este compuesto alcanzada en el líquido cefalorraquídeo que está muy lejos de la necesaria para causar neurotoxicidad in vitro95,96.
También se ha propuesto que no fuese una acción propia, sino que la HCY pudiera potenciar las lesiones producidas por otras causas como las producidas mediante la inyección de kainato95, la mutación de la proteína precursora de amiloide que se da en ratones transgénicos para esta proteína, que desarrollan enfermedad de Alzheimer97, o en la inducción farmacológica de la enfermedad de Parkinson98.
Homocisteína y otras patologíasLa HHCY también es un factor independiente en fracturas osteoporóticas en ancianos y está asociada con la disminución de la densidad ósea99–101. Además, está relacionada de manera directa con la presión arterial; se ha determinado que un aumento de 5μM está asociado con un incremento de 0,5 y de 0,7mmHg en la presión sistólica de hombres y mujeres, respectivamente102. La HHCY puede alterar la configuración de la retina por cambios transcripcionales103. Y, por último, se ha encontrado relación entre los niveles de HCY y la resistencia a la insulina en pacientes obesos no diabéticos104. En esta última acción parece estar implicada una inducción de la secreción de resistina adipocitaria por parte de la HCY mediada por aumento de estrés oxidativo, proteína quinasa C y NF-κB105.
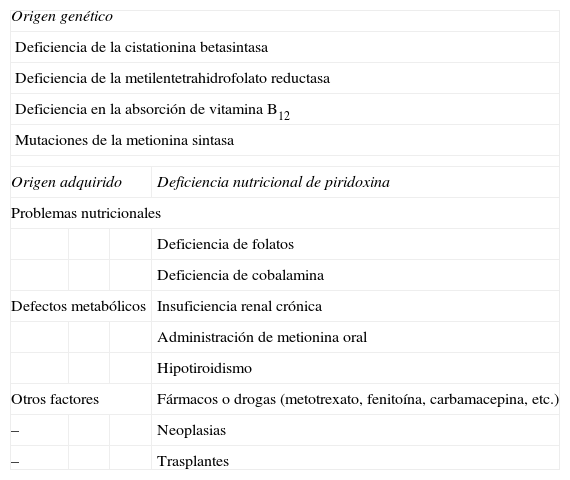
Etiología de la hiperhomocisteinemiaLa HHCY puede ser congénita o adquirida, y esta a su vez puede ser de origen multifactorial. Estas posibilidades se resumen en la tabla 1.
Etiología de la hiperhomocisteinemia
| Origen genético | |||
| Deficiencia de la cistationina betasintasa | |||
| Deficiencia de la metilentetrahidrofolato reductasa | |||
| Deficiencia en la absorción de vitamina B12 | |||
| Mutaciones de la metionina sintasa | |||
| Origen adquirido | Deficiencia nutricional de piridoxina | ||
| Problemas nutricionales | |||
| Deficiencia de folatos | |||
| Deficiencia de cobalamina | |||
| Defectos metabólicos | Insuficiencia renal crónica | ||
| Administración de metionina oral | |||
| Hipotiroidismo | |||
| Otros factores | Fármacos o drogas (metotrexato, fenitoína, carbamacepina, etc.) | ||
| – | Neoplasias | ||
| – | Trasplantes | ||
La causa congénita más frecuente es el defecto de la CBS, cuyo gen se halla ubicado en el cromosoma 21106,107. El defecto se transmite de forma autosómica recesiva. La deficiencia de CBS en homocigosis tiene una incidencia de entre 1:58.000–1:1.000.000, dependiendo del país estudiado8,108. En esta situación, los pacientes pueden llegar a tener valores de HCY plasmática de hasta 400μmol/l. La frecuencia del alelo heterocigoto aumenta considerablemente en la población, pudiendo llegar hasta el 1%109.
Otro defecto genético muy común asociado a una moderada HHCY es la mutación puntual en la región codificante del gen de la metilentetrahidrofolato reductasa (MTHFR). Esta enzima está asociada a la remetilación de la HCY. La mutación genera una variante termolábil de la enzima que tiene disminuida su actividad en un 50%110,111.
La insuficiencia renal o las deficiencias en vitaminas B requeridas por el metabolismo de la HCY, como el ácido fólico, la vitamina B6 y/o la vitamina B12, también son causa de HHCY112,113. Se estima que una inadecuada ingesta de estas vitaminas es la responsable de 2/3 de todos los casos de HHCY114,115. Aunque la deficiencia de folato y vitamina B12 están relacionados con un aumento de la concentración de la HCY plasmática116,117, la relación entre la HCY y los niveles de vitamina B6 está menos clara118. Consecuentemente, los fármacos que interfieren en el metabolismo de estas vitaminas también pueden producir HHCY119–121. Además, se ha observado recientemente que un exceso de metionina en la dieta produce HHCY en los ratones60.
También se han obtenido asociaciones entre los niveles de HCY y otros hábitos característicos que conforman nuestro estilo de vida. El estudio Hordaland es el primero a gran escala que refleja que el tabaco, el abundante consumo de café y la falta de ejercicio están asociados con concentraciones elevadas de HCY122–125. Los factores genéticos afectan mayoritariamente a los niveles de HCY de la población joven. Mientras que en los ancianos estos efectos tienen una repercusión considerablemente más pequeña que los introducidos por los factores nutricionales126.
FisiopatologíaHiperhomocisteinemia y disfunción endotelialComo se ha comentado anteriormente, la HHCY es un factor de riesgo independiente en el desarrollo de enfermedades cardiovasculares; sin embargo, los mecanismos de acción no son conocidos. La mayoría de los experimentos apoyan la hipótesis de que el exceso de HCY conlleva una disfunción endotelial y, como se ha observado anteriormente, esta disfunción sería crucial en la patogénesis y el desarrollo de la arteriosclerosis, tal como se resume en la tabla 2. Dentro de esta disfunción se pueden destacar distintos mecanismos por los que la HHCY afectaría al endotelio.
Resumen de los mecanismos propuestos para la acción de la homocisteína
| Arquitectura de los vasos | Estrés oxidativo | ↑ | |
| Daño endotelial | ↑ | Producción de peroxinitritos | ↑ |
| Proliferación de VSMC | ↑ | Enzimas oxidativas (SOD, GPx) | ↑ |
| Síntesis de colágeno y fibrosis de la media | ↑ | Peroxidación lipídica | ↑ |
| Restenosis vascular | ↑ | Quimiotaxis, adhesión de leucocitos | ↑ |
| Formación de células espumosas | ↑ | Adhesión de leucocitos | ↑ |
| Placas fibroproliferativas | ↑ | Presencia de ICAM-1, VCAM-1 | ↑ |
| Daño en estructura celular | ↑ | Quimiotaxis (IL-8, MCP-1) | ↑ |
| Daños mitocondriales | ↑ | Activación de la coagulación | ↑ |
| Estrés del retículo endoplásmico | ↑ | Factor tisular | ↑ |
| Metaloproteinasas | ↑ | Inactivación de la proteína C | ↑ |
| Elastólisis | ↑ | Complejo trombina-antitrombina | ↑ |
| Expresión de HSP70 | ↓ | D-Dímer | ↑ |
| Disfunción endotelial | ↑ | Fibrinólisis | ↓ |
| Sistema óxido nítrico (NO) | ↓↑ | Sulfato de heparina | ↓ |
| Biodisponibilidad de NO | ↓ | Anexina II | ↓ |
| ADMA | ↑ | Trombomodulina | ↓ |
| Factores de transcripción | Antígenos PAI-1, t-PA | ↑ | |
| Activación de NF-κB, SREBP, PKC | ↑ | Fragmento de protrombina | ↑ |
| Expresión de los genes | ↓↑ | Inactivación del factor Va | ↓ |
| HMG-CoA reductasa | ↑ | Agregación trombocitaria | ↑ |
| Biosíntesis de lípidos | ↑ | Función de fibronectina | ↓ |
| Inactivación de PPAR alfa y gamma | ↑ | COX, producción de TXA, TXB | ↑ |
ADMA: dimetilarginina asimétrica; COX: ciclooxigenasa; Gpx: glutatión peroxidasa; IL-8: interleucina 8; NO: óxido nítrico; SOD: superóxido dismutasa; t-PA: activador del plasminógeno tisular; TXA: tromboxano A; TXB: tromboxano B.
Diferentes estudios realizados tanto en humanos como en modelos animales han demostrado que la HHCY induce una deficiente vasodilatación de las arterias127–129. Esta alteración produce daños similares a los provocados por otros factores de riesgo, como la hipercolesterolemia y la hipertensión130. Stamler et al demostraron que las propiedades vasodilatadoras de la célula endotelial se veían disminuidas por la HCY debido principalmente a una reducción en la producción de óxido nítrico (NO)131. La inactivación del NO por la HCY se ha observado en cultivos de células endoteliales e in vivo27,128, y tiene lugar por una reducción de la expresión proteica de la eNOS y una disminución de su actividad por fosforilación en S1179132.
Algunas especies reactivas de hidrógeno, como el superóxido, el peróxido de hidrógeno y los peroxinitritos, pueden contribuir a la inactivación del NO derivado del endotelio133. Otro mecanismo que puede actuar en este sentido es la inhibición de la producción de NO causada por la dimetilarginina asimétrica (ADMA). La ADMA es un compuesto considerado como el inhibidor fisiológico de la NO sintasa y que se origina en la hidrólisis de proteínas metiladas. Se observan niveles plasmáticos elevados de ADMA en primates con HHCY134,135. En pacientes sanos, los niveles de ADMA aumentan rápidamente después de una sobrecarga de metionina y se correlacionan con una alterada vasodilatación dependiente del endotelio136. Este acúmulo se produce por la inhibición de la enzima dimetilarginina dimetilamino hidrolasa responsable de la eliminación de la ADMA137.
La HCY también ejerce una acción directa sobre los miocitos, donde activa la metaloproteinasa 9 e induce permeabilidad mitocondrial y disfunción mecánica de este tipo de células. Se ha propuesto que esta acción podría estar mediada por una interacción de la HCY con el receptor de N-metil-D-aspartato-1138,139 y ejercerse a través de la señalización del ácido epoxieicosatrienoico140 y de las proteínas G141 e independiente de la NO sintasa inducible142.
Estrés oxidativoVarias líneas de investigación sugieren que el incremento del estrés oxidativo y de los niveles de especies reactivas de oxígeno desempeñan un papel importante en los cambios vasculares producidos en una situación de HHCY128,143,144. La primera hipótesis que surge de estas observaciones es que la HCY autooxida su grupo tiol altamente reactivo y, a su vez, forma especies reactivas de oxígeno. Sin embargo, esta hipótesis no puede explicar cómo la cisteína, que se encuentra en cantidades superiores a la HCY y también se puede autooxidar, no causa daño endotelial y no es considerada como factor de riesgo en las enfermedades cardiovasculares.
El estrés oxidativo generado por la HCY puede afectar a la aterogénesis por mecanismos distintos a la autooxidación; por ejemplo, un exceso de HCY puede reaccionar con el NO para formar la S-nitrosohomocisteína, impidiendo así una correcta respuesta del endotelio. También se ha descrito que la HHCY produce un descenso de la expresión de la glutatión peroxidasa (GPx), lo que impediría la inactivación del ión superóxido, de forma que ese ión podría reaccionar con el NO y formar peroxinitrito. La consecuencia, igualmente, sería la menor disponibilidad del NO para la vasodilatación145.
Otro dato para tener en cuenta es que la HCY puede incrementar el estrés oxidativo inhibiendo la expresión de la enzima superóxido dismutasa, produciendo de esta manera un aumento del anión superóxido y de los peroxinitritos98,146. Tanto el anión superóxido como los peroxinitritos contribuyen a la generación de peróxidos lipídicos147 y a la modificación de proteínas mediante la nitración del aminoácido tirosina. El peroxinitrito puede ejercer varios efectos; el primero es la nitración en tirosina de la prostaciclina sintasa y la superóxido dismutasa, la isoforma principal en la mitocondria, además de la activación de la poli-ADP ribosa polimerasa, importantes mediadores de la disfunción vascular148. La inhibición de esta enzima atenúa el desarrollo aterosclerótico inducido por la HHCY a través de la inhibición del NF-κB149. También puede impedir la función endotelial oxidando el cofactor de la eNOS, la tetrahidrobiopterina, produciendo de esta manera una disminución en su actividad y/o el desacoplamiento de la enzima150. En este sentido, existen resultados que indican que la expresión y la actividad de la GPx y de la HO-1 están disminuidas en cultivos de células del endotelio vascular en situación de HHCY, lo que sugiere que la HCY puede inhibir el potencial antioxidante de las células151. Además, cuando al ratón deficiente en GPx-1 se le provocó una HHCY moderada con dietas deficientes en folato, se confirmó que la disfunción endotelial anteriormente observada en ratones con HHCY moderada se exacerbaba con la deficiencia añadida de GPx-1151 y que la sobreexpresión de esta enzima en animales carentes de una copia del gen CBS recuperaba la disfunción27. Ambos resultados apuntan inequívocamente al papel de los peróxidos en el desarrollo de la disfunción endotelial ocasionada por la HHCY.
Esta generación de radicales libres podría incrementar la oxidación de las lipoproteínas de baja densidad (LDL) y, por lo tanto, su captación por parte de los macrófagos en la pared vascular152. Más recientemente se han obtenido resultados que apoyan esta hipótesis; Nakano et al observaron la oxidación de LDL humanas mediada por la HCY. Las LDL oxidadas mediante este procedimiento son capturadas rápidamente por los macrófagos a través de la activación del receptor scavenger. Estos resultados indican que la salida continua de HCY desde las células endoteliales contribuye a la modificación de las LDL y a su incorporación al macrófago, un paso muy importante en el desarrollo de la arteriosclerosis153.
InflamaciónSe ha puesto de manifiesto que en una situación de HHCY se induce la producción de diversas moléculas proinflamatorias. En este sentido, se ha demostrado que el tratamiento de células del endotelio vascular, de células del músculo liso y de monocitos con HCY induce la expresión de MCP-1, IL-8, VCAM-1, Selectina E154 y CXCL16 y de su receptor CXCR6155. MCP-1 aumenta la unión de los monocitos al endotelio y su reclutamiento hacia el espacio subendotelial, un paso crítico en el desarrollo de la lesión aterosclerótica. Además, la inducción de IL-8 y MCP-1 se produce a través de la activación de NF-κB, un factor de transcripción que estimula la producción de citoquinas, moléculas de adhesión y factores de crecimiento que, a su vez, contribuyen a la inflamación vascular156. Por último, la HCY incrementa la expresión de TNF-α que conlleva una estimulación de otros genes inflamatorios y la inhibición de la vasoconstricción con la consiguiente alteración de la función endotelial157. La HCY promueve el estrés oxidativo en monocitos a través de la NAD(P)H oxidasa158, la disminución del glutatión159 o alteraciones en la tioredoxina160.
CoagulaciónLa función del endotelio en el control de la coagulación ha sido abordada mediante cultivos celulares. Así, Rodgers et al demostraron que las células endoteliales en presencia de HCY aumentan la expresión del factor v y la activación de la protrombina, lo que favorecería un estatus procoagulante161. Por otra parte, la HCY inhibe también importantes vías fisiológicas anticoagulantes, como son la activación de la proteína C y la expresión de la trombomodulina en la superficie endotelial162,163. Además, se han encontrado alteraciones en la unión con la antitrombina iii164 y la reducción de la unión del activador tisular del plasminógeno a su receptor endotelial165, un aumento en la secreción del factor Von Willebrand166 y un incremento de la expresión del factor tisular por parte del endotelio expuesto a la HCY167. Por último, se ha observado que en conejos con HHCY inducida se producen coágulos anormalmente resistentes a la fibrinólisis que podrían contribuir directamente al proceso de trombosis168 y, mediante inhibición de la fibrinólisis, impedir la migración de células endoteliales169.
Otros mecanismos implicadosAunque los recientes trabajos experimentales se centran en el NO y el estrés oxidativo, hay que poner de manifiesto que la HCY tiene otras acciones que pueden alterar el endotelio. La HCY produce la hipometilación del DNA170 y altera la regulación por metilación de proteínas como Ras171. Igualmente, se ha propuesto un estrés del retículo172–174; sin embargo, nuevos estudios parecen sugerir que este fenómeno sería una consecuencia más que un efecto directo175. Además, se ha descubierto que participa como aminoácido proteinogénico al ser la S-NO-HCY análoga a la metionina e incorporarse al tARNmet. La incorporación de estos residuos en diferentes proteínas podría inducir un grave daño en las células176,177 y es un hallazgo corroborado tanto en humanos como en ratones178,179. La HCY induce también la proliferación de las células musculares lisas y disminuye la síntesis del DNA endotelial180,181. Todos estos efectos pueden desembocar en una alteración de la actividad vascular y, en última instancia, en la activación de muerte celular en el endotelio182.
Modelos animales de hiperhomocisteinemiaDada la relevancia de la elevación de la HCY en la patología y la necesidad de profundizar cada vez más en los mecanismos, se hacen necesarios nuevos desarrollos experimentales más rigurosos y eficaces para avanzar en el conocimiento. En este sentido, el ratón es un instrumento muy valioso debido a la variedad de modelos modificados genéticamente disponibles, su facilidad de manejo, el desarrollo de técnicas de alta sensibilidad muy útiles para muestras pequeñas y las consideraciones económicas, que hacen del ratón el modelo animal más utilizado.
Las estrategias posibles para generar HHCY quedan reflejadas en la figura 3 y se enfocan a reducir la actividad de las enzimas implicadas en el metabolismo tanto nutricional como genéticamente, tal como desarrollamos a continuación.

Estrategias de modificación del metabolismo de la homocisteína para lograr hiperhomocisteinemia. BHMT: betaína homocisteína metiltransferasa; CBS: cistationina betasintasa; Metil THF: metiltetrahidrofolato; Metilen THF: metilentetrahidrofolato; MS: metionina sintasa; MSR: metionina sintasa reductasa; MTHFR: metilentetrahidrofolato reductasa; SAH: S-adenosil homocisteína hidrolasa; SAM: S-adenosil metionina; SMMT: S-metil metionina metiltransferasa; THF: tetrahidrofolato.
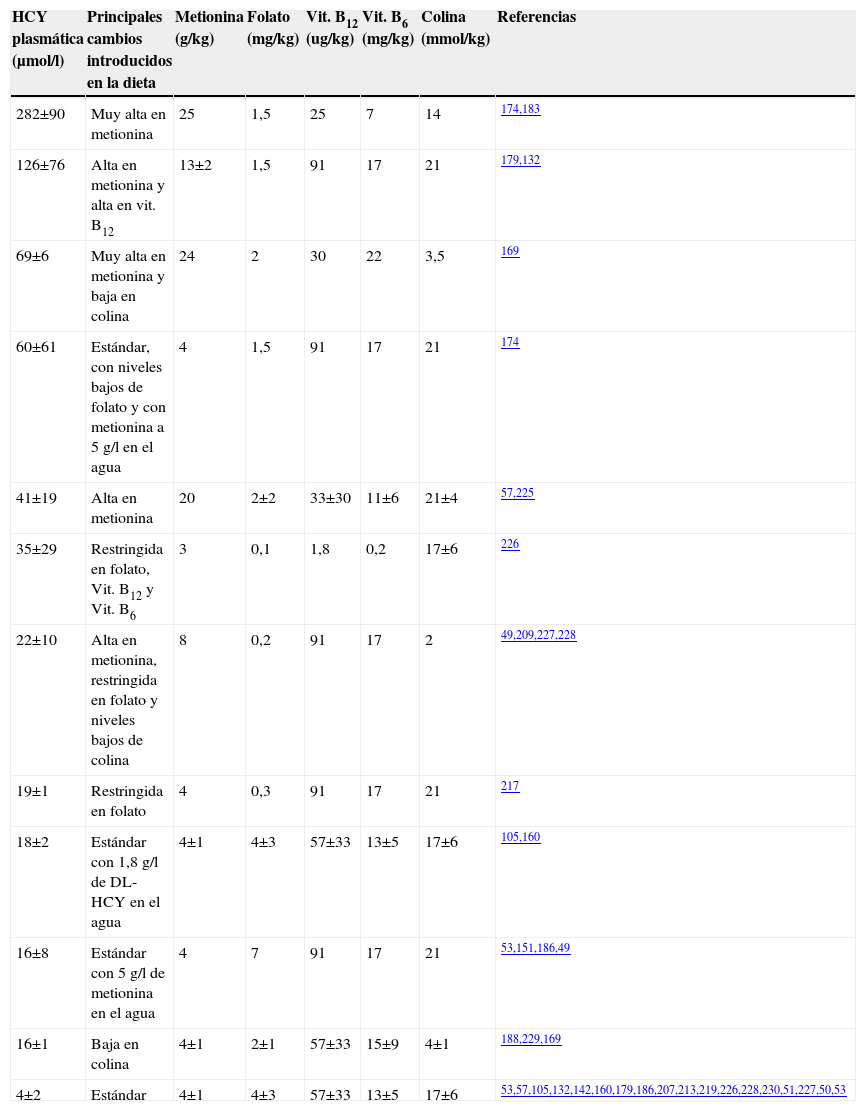
La tablas 3 y 4 recogen algunas de las estrategias llevadas a cabo in vivo con ratones de diferentes estirpes presentes en la naturaleza. La elevación de HCY en el plasma se puede inducir a través de varias vías: la más agresiva sería el aumento de metionina hasta alcanzar los 25g/kg y la disminución de los aportes de folato, vitamina B12, B6 y colina en la dieta174,183. La cepa de ratón C57Bl/6J responde a estas dietas de forma bastante más acusada cuando se compara con la raza BALB/c. Tras un incremento inicial, se observó que las concentraciones de HCY en el plasma disminuían. En algunos casos, estos experimentos se han prolongado hasta un año y con este tipo de dietas se consiguieron niveles de HCY en el plasma superiores a 240μmol/l, lo que se aproxima mucho a los niveles que se pueden obtener trabajando con animales modificados genéticamente. Es de destacar que en animales alimentados con pienso alto en metionina y bajo en folato durante 8 meses se han observado alteraciones como esteatosis hepática y niveles de metionina en el plasma elevados, efectos que podrían alterar los resultados de los estudios. Esto podría ser explicado porque esas cantidades de metionina en el pienso son tóxicas y, además, pueden ocasionar retrasos en el crecimiento.
Estrategias dietéticas utilizadas en ratones de la cepa C57BL/6J y las concentraciones de homocisteína total conseguidas en plasma. Adaptada de (224)
| HCY plasmática (μmol/l) | Principales cambios introducidos en la dieta | Metionina (g/kg) | Folato (mg/kg) | Vit. B12 (ug/kg) | Vit. B6 (mg/kg) | Colina (mmol/kg) | Referencias |
| 282±90 | Muy alta en metionina | 25 | 1,5 | 25 | 7 | 14 | 174,183 |
| 126±76 | Alta en metionina y alta en vit. B12 | 13±2 | 1,5 | 91 | 17 | 21 | 179,132 |
| 69±6 | Muy alta en metionina y baja en colina | 24 | 2 | 30 | 22 | 3,5 | 169 |
| 60±61 | Estándar, con niveles bajos de folato y con metionina a 5g/l en el agua | 4 | 1,5 | 91 | 17 | 21 | 174 |
| 41±19 | Alta en metionina | 20 | 2±2 | 33±30 | 11±6 | 21±4 | 57,225 |
| 35±29 | Restringida en folato, Vit. B12 y Vit. B6 | 3 | 0,1 | 1,8 | 0,2 | 17±6 | 226 |
| 22±10 | Alta en metionina, restringida en folato y niveles bajos de colina | 8 | 0,2 | 91 | 17 | 2 | 49,209,227,228 |
| 19±1 | Restringida en folato | 4 | 0,3 | 91 | 17 | 21 | 217 |
| 18±2 | Estándar con 1,8g/l de DL-HCY en el agua | 4±1 | 4±3 | 57±33 | 13±5 | 17±6 | 105,160 |
| 16±8 | Estándar con 5g/l de metionina en el agua | 4 | 7 | 91 | 17 | 21 | 53,151,186,49 |
| 16±1 | Baja en colina | 4±1 | 2±1 | 57±33 | 15±9 | 4±1 | 188,229,169 |
| 4±2 | Estándar | 4±1 | 4±3 | 57±33 | 13±5 | 17±6 | 53,57,105,132,142,160,179,186,207,213,219,226,228,230,51,227,50,53 |
HCY: homocisteína; Vit.: vitamina.
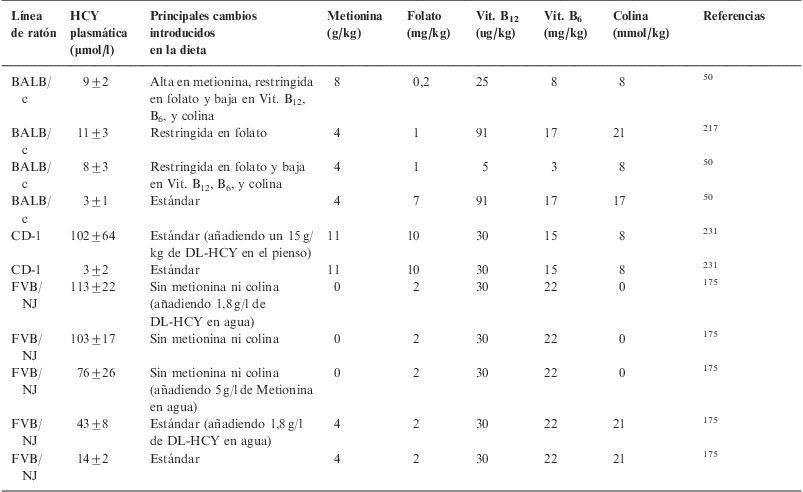
Estrategias dietéticas utilizadas para provocar hiperhomocisteinemia en otras razas de ratones
| Línea de ratón | HCY plasmática (μmol/l) | Principales cambios introducidos en la dieta | Metionina (g/kg) | Folato (mg/kg) | Vit. B12 (ug/kg) | Vit. B6 (mg/kg) | Colina (mmol/kg) | Referencias |
| BALB/c | 9±2 | Alta en metionina, restringida en folato y baja en Vit. B12, B6, y colina | 8 | 0,2 | 25 | 8 | 8 | 50 |
| BALB/c | 11±3 | Restringida en folato | 4 | 1 | 91 | 17 | 21 | 217 |
| BALB/c | 8±3 | Restringida en folato y baja en Vit. B12, B6, y colina | 4 | 1 | 5 | 3 | 8 | 50 |
| BALB/c | 3±1 | Estándar | 4 | 7 | 91 | 17 | 17 | 50 |
| CD-1 | 102±64 | Estándar (añadiendo un 15g/kg de DL-HCY en el pienso) | 11 | 10 | 30 | 15 | 8 | 231 |
| CD-1 | 3±2 | Estándar | 11 | 10 | 30 | 15 | 8 | 231 |
| FVB/NJ | 113±22 | Sin metionina ni colina (añadiendo 1,8g/l de DL-HCY en agua) | 0 | 2 | 30 | 22 | 0 | 175 |
| FVB/NJ | 103±17 | Sin metionina ni colina | 0 | 2 | 30 | 22 | 0 | 175 |
| FVB/NJ | 76±26 | Sin metionina ni colina (añadiendo 5g/l de Metionina en agua) | 0 | 2 | 30 | 22 | 0 | 175 |
| FVB/NJ | 43±8 | Estándar (añadiendo 1,8g/l de DL-HCY en agua) | 4 | 2 | 30 | 22 | 21 | 175 |
| FVB/NJ | 14±2 | Estándar | 4 | 2 | 30 | 22 | 21 | 175 |
HCY: homocisteína; Vit.: vitamina.
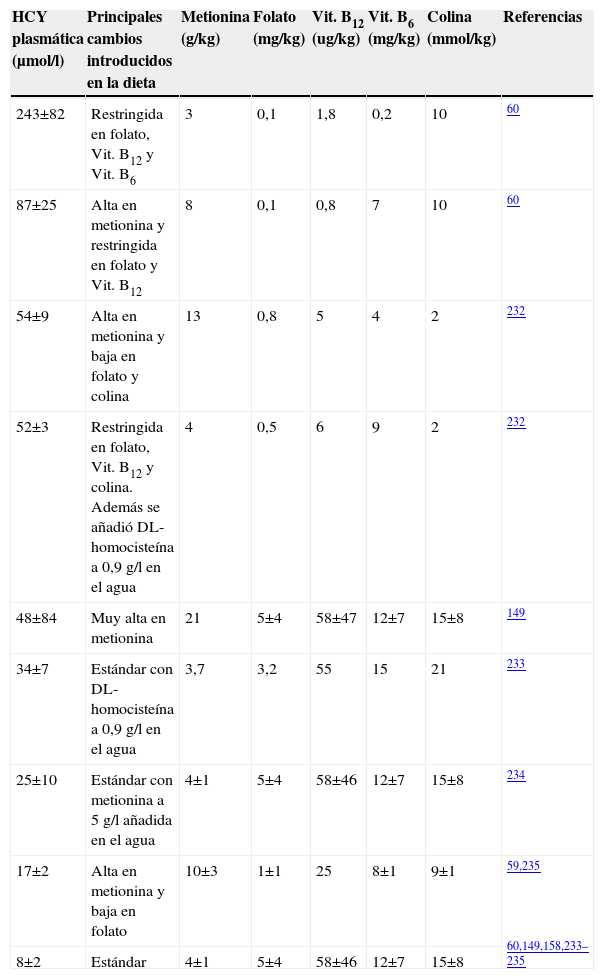
En el caso de los ratones modificados genéticamente, se han utilizado los deficientes en ApoE (tabla 5) a los que se les indujo HHCY mediante el aporte extra de metionina en la dieta y la eliminación de las vitaminas B12, B6 y ácido fólico. Con este enfoque experimental, Hofmann et al encontraron que la HHCY inducía un incremento en la expresión y actividad de factores clave en procesos inflamatorios, aterogénesis y vulnerabilidad de las placas aterogénicas, tales como la molécula de adhesión de células vasculares, el factor tisular y la metaloproteinasa 958.
Estrategias dietéticas utilizadas para modificar la homocisteinemia en ratones carentes de apolipoproteína E
| HCY plasmática (μmol/l) | Principales cambios introducidos en la dieta | Metionina (g/kg) | Folato (mg/kg) | Vit. B12 (ug/kg) | Vit. B6 (mg/kg) | Colina (mmol/kg) | Referencias |
| 243±82 | Restringida en folato, Vit. B12 y Vit. B6 | 3 | 0,1 | 1,8 | 0,2 | 10 | 60 |
| 87±25 | Alta en metionina y restringida en folato y Vit. B12 | 8 | 0,1 | 0,8 | 7 | 10 | 60 |
| 54±9 | Alta en metionina y baja en folato y colina | 13 | 0,8 | 5 | 4 | 2 | 232 |
| 52±3 | Restringida en folato, Vit. B12 y colina. Además se añadió DL-homocisteína a 0,9g/l en el agua | 4 | 0,5 | 6 | 9 | 2 | 232 |
| 48±84 | Muy alta en metionina | 21 | 5±4 | 58±47 | 12±7 | 15±8 | 149 |
| 34±7 | Estándar con DL-homocisteína a 0,9g/l en el agua | 3,7 | 3,2 | 55 | 15 | 21 | 233 |
| 25±10 | Estándar con metionina a 5g/l añadida en el agua | 4±1 | 5±4 | 58±46 | 12±7 | 15±8 | 234 |
| 17±2 | Alta en metionina y baja en folato | 10±3 | 1±1 | 25 | 8±1 | 9±1 | 59,235 |
| 8±2 | Estándar | 4±1 | 5±4 | 58±46 | 12±7 | 15±8 | 60,149,158,233–235 |
HCY: homocisteína; Vit.: vitamina.
Estos efectos revertían si se eliminaba la HHCY. También se observa un aumento de tres factores de transcripción (SREBP-2, CREB y NF-Y) en el hígado de ratas con HHCY inducida en la dieta. Estos efectos se asocian al incremento de la actividad de la hidroximetil glutaril coenzima A reductasa que conlleva el acúmulo de lípidos en el hígado184. Trabajos más recientes han suscitado controversia sobre este enfoque, ya que la HHCY inducida por aporte de metionina en la dieta no contribuyó a la progresión de la aterosclerosis en ratones carentes de ApoE185. Un efecto que también ha sido observado en ratones C57BL/6186 a pesar de los cambios proinflamatorios y el aumento de lípidos plasmáticos.
Otro posible tipo de intervención encaminado a lograr HHCY a través de la dieta se ha centrado en reducir únicamente las cantidades de folato y vitaminas B12 y B6, tal como se observa en la tabla 3. Con este desarrollo experimental se podían obtener concentraciones de 10–20μmol/l, lo que imita la situación crónica humana. Los resultados demostraron alteraciones en las arteriolas del cerebelo o en las mesentéricas, necesitándose estrategias más agresivas con concentraciones por encima de 20μmol/l para conseguir lesiones en los vasos mayores, como la aorta o la carótida47,48,50.
La última forma de inducir HHCY ha sido la adición de HCY directamente en el agua de bebida combinándola en algunos casos con dietas especiales carentes de las vitaminas implicadas en el metabolismo de metionina y HCY. En esta situación, los valores de hiperhomocisteína también fueron moderados (tablas 3–5).
La manipulación de la dieta durante largos periodos de tiempo permite modular la cantidad de HCY que se desea obtener en el plasma y así emular la situación crónica presente en pacientes humanos. Otra ventaja potencial de la inducción de HHCY por vía dietética sería la posibilidad de trabajar con animales no transgénicos, con lo que se evitarían los problemas reproductivos que pueden presentar las diferentes líneas de ratones modificados genéticamente, comentados más adelante, y que condicionan los diseños experimentales. Por otro lado, este tipo de experimentación plantea una serie de inconvenientes: el primero, la toma de muestras, ya que se ha demostrado que los niveles de HCY son mucho mayores en animales recién alimentados que en los ayunados187, por ello se han de controlar estrictamente los tiempos de ayuno y esto exige un gran esfuerzo y la utilización de jaulas metabólicas de última generación. Otro problema encontrado en los diversos estudios dietéticos se refiere al uso de piensos control semipurificados, cuya fuente de proteína (soja, maíz, caseína) puede variar en función de los vaivenes del mercado, por ello se hace cada vez más imperante el empleo de dietas purificadas cuya fuente de proteína sea constante. Por último, uno de tipo conceptual, ya que la mayor limitación del uso de dietas para inducir HHCY es que, al utilizar dietas deficientes en algunas sustancias esenciales, se podrían estar modificando las lesiones que observamos, de forma independiente de las que provoque la HCY per se.
Inducción de hiperhomocisteinemia mediante tratamiento farmacológico en ratonesS-(δ-carboxilbutil)-DL-HCY es un inhibidor de la betaína HCY metiltransferasa que, inyectado intraperitonealmente durante tres días, provoca unos aumentos de HCY plasmáticas de hasta unos 18μmol/l188. Otro sistema utilizado es la implantación subcutánea en ratones de bombas osmóticas que liberen HCY de forma lenta y sostenida en el tiempo189. Los inconvenientes de esta técnica son la necesidad de anestesia y el procedimiento quirúrgico, y la ventaja es la posibilidad de controlar la HCY plasmática.
Modelos genéticos de hiperhomocisteinemia e hipohomocisteinemiaAl objeto de disponer de modelos animales con HHCY permanente, se han generado por ingeniería genética diversos ratones carentes de las enzimas MS, metionina sintasa reductasa, MTHFR y CBS.
Los ratones heterocigotos carentes del gen de la MS fueron moderadamente hiperhomocisteinémicos, en tanto que los embriones homocigotos murieron poco después de la implantación, lo que sugiere un papel crucial de esta enzima en el desarrollo embrionario de este animal190. Mediante el empleo de estos animales se ha observado incapacidad de respuesta vascular y aumento de estrés oxidativo48.
Los ratones homocigotos carentes de metionina sintasa reductasa, que es la enzima que cataliza la reducción de la MS, no poseen problemas de letalidad o de crecimiento, ya que no tienen totalmente perdida la actividad de esta enzima, sino que tienen disminuida su expresión génica entre un 1–37% comparados con un animal normal. Presentan una leve HHCY (18μmol/l frente a 5μmol/l del control) pero tienen la metionina elevada en un 30%191.
Los ratones carentes de la enzima MTHFR se generaron por dos grupos de manera independiente192,193. La enzima MTHFR reduce este compuesto a 5-metiltetrahidrofolato, que se utiliza en la remetilación de la HCY. El fallo de esta enzima es frecuente en la población, por lo que es interesante el estudio del animal carente de dicha enzima. Los ratones homocigotos deficientes en la enzima MTHFR presentaron unos niveles de HCY 10 veces superiores a los controles, en tanto que la inactivación de la enzima en heterocigosis solo elevó dicho parámetro 1,6 veces el valor normal. Los animales homocigotos presentaron menor tamaño corporal y retraso del desarrollo, así como alteraciones patológicas en el cerebelo, en concreto, disminución de su tamaño y de la presencia de células granulosas, aunque no la neurogénesis194. En estos animales, tanto en heterocigosis como en homocigosis, aunque a una edad avanzada, se observaron depósitos de lípidos en la aorta192. En este modelo animal se encontró igualmente una correlación negativa significativa entre la betaína plasmática y las concentraciones de HCY. Se presentó, asimismo, una disminución en la concentración hepática de betaína, dimetilglicina y otros metabolitos de la colina, lo que sugiere que la disminución o la ausencia de la MTHFR alteran la homeostasis de la colina. Estos cambios metabólicos se reflejaron en una severa esteatosis hepática. El suplemento de betaína en la dieta mejoró la supervivencia y el crecimiento de los animales homocigotos, redujo la HCY plasmática, aumentó la ApoA-I y revirtió algunos de los cambios patológicos observados, tales como las anomalías cerebelosas188, la espermatogénesis defectuosa195 y los acúmulos lipídicos del hígado y de la aorta196. Estos resultados evidencian la extraordinaria sensibilidad de estos modelos a los cambios de betaína de la dieta.
En este mismo tipo de animales, también se observó una hipometilación del DNA así como un descenso de la S-adenosil metionina y un aumento de la S-adenosil HCY que podrían explicar el efecto observado a nivel del DNA. La disminución de adenosina puede contribuir a los efectos cardiovasculares de la HHCY. In vivo, cualquier incremento de HCY en el plasma refleja un incremento intracelular de HCY que, inevitablemente, disminuye la adenosina debido a que se acumula la adenosil HCY. De este modo se perdería la acción beneficiosa de la estimulación de los receptores de adenosina que produce varias acciones cardioprotectoras, la vasodilatación197, la inhibición de la agregación plaquetaria198, la modulación de la inflamación y la regulación vascular de la proliferación y la muerte celular50,199,200.
El primer modelo genético de HHCY, creado en 1995 por el grupo de Maeda, fue el ratón carente de la enzima CBS71. Los ratones carentes de esta enzima presentaron una HHCY muy severa y completaron el desarrollo embrionario normalmente71. Los animales neonatos presentan alteraciones del crecimiento reflejadas en un retraso de la apertura del ojo y anormalidades esqueléticas tales como la cara, la cola y las extremidades muy alargadas y delgadas. El 80% de estos ratones muere hacia el día 21 de su nacimiento. Las investigaciones de las anormalidades morfológicas revelan que estos ratones poseen la piel arrugada, la dermis y la hipodermis muy finas y sufren de hiperqueratosis de la epidermis. El pelo en el lomo es menos denso y con menor diámetro que los animales control. Estos resultados demuestran que las anormalidades cutáneas observadas en el ratón deficiente en CBS se asemejan a las encontradas en humanos y que están relacionadas con la homeostasis de la matriz extracelular91. Recientemente, se ha observado que en estos animales existe un acortamiento de los huesos largos debido a una diferenciación deficiente en el cartílago201. El aporte de betaína a ratones heterocigotos del gen de CBS redujo la concentración de HCY188 a pesar de que, en este caso, el suplemento de cisteína resulta especialmente crítico para su supervivencia202. En este tipo de animales, el aporte de vino tinto redujo los niveles de HCY y mejoró los parámetros de función endotelial203.
Un importante hallazgo fue la histopatología hepática. Los hepatocitos eran de mayor tamaño del normal, multinucleados o binucleados y con inclusiones lipídicas intracitoplasmáticas. El análisis de lípidos reveló acúmulo de colesterol y triglicéridos en este órgano, en consonancia con los hallazgos histopatológicos. En el origen de dicho problema se postuló una exacerbada captación de lípidos plasmáticos por una desregulación de la ruta de respuesta a esteroles a consecuencia del estrés del retículo endoplásmico174. La HHCY severa de este modelo también se acompañó de hipometilación del DNA y elevación de la S-adenosil HCY en el hígado204. Igualmente, los machos que sobrevivieron fueron fértiles, mientras que las hembras no lo fueron71. En la investigación de las causas se comprobó que no se debía a problemas directamente del feto porque se obtenían animales homocigotos vivos nacidos de hembras heterocigotas. Por el contrario, las hembras homocigotas carentes del gen no llegaban a parir crías vivas, debiéndose a un problema en el útero, demostrado al realizar trasplantes de ovarios de hembras homocigotas carentes de CBS a receptoras normales que se cruzaron con machos homocigotos. Estas receptoras, en cambio, fueron capaces de parir animales vivos homocigotos carentes del gen72.
Al estudiar la expresión de los genes hepáticos de ratones deficientes en CBS, se observan cambios en genes implicados en la proliferación y en el crecimiento celular. También están alterados algunos genes del estrés oxidativo, por ejemplo, se sobreexpresa la HO-1 y disminuye la expresión de la paraoxonasa205. Recientemente se ha demostrado que la HHCY severa promueve en el hígado estrés oxidativo, que puede causar daño mitocondrial en asociación con la activación de células hepáticas esteladas206. Sin embargo, existen señales protectoras que contrarrestan las señales apoptóticas en el hígado de estos ratones, tales como el aumento de la actividad catalasa207 y de la activación de la degradación del inhibidor κB por la calpaína208. Otros estudios en el hígado demuestran que los ratones deficientes en CBS tienen alterada la homeostasis redox con niveles de glutatión y cisteína reducidos. Por el contrario, los heterocigotos solo presentan mínimos cambios en el estado redox y no muestran ninguna desventaja frente a los controles209.
En los cerebros de los ratones deficientes en CBS, algunos genes de la vía de las quinasas SAPK/JNK se encuentran alterados. Investigando la activación de proteínas involucradas en esta cascada, se ha demostrado que JNK y c-Jun están activadas en neuronas del hipocampo, sugiriendo que esta vía puede desempeñar un papel importante en el desarrollo de defectos neuronales asociados a la HHCY91.
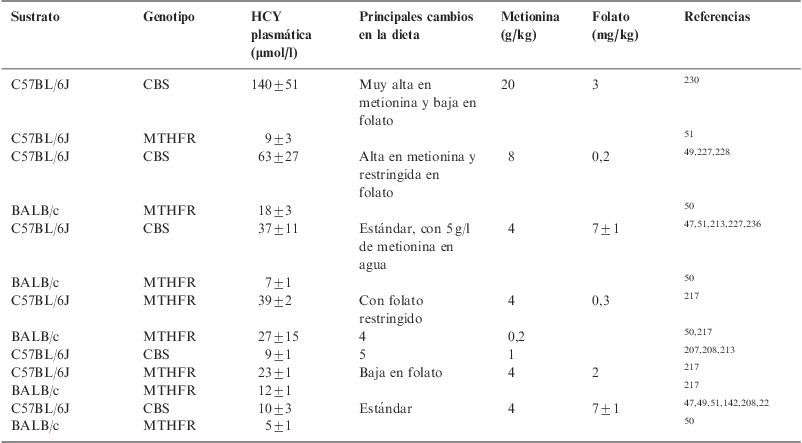
En estos modelos de HHCY, la modificación dietética también permite manipular la intensidad de esta, tal como se recoge en la tabla 6 para los ratones heterocigotos carentes de CBS y MTHFR. Con excepción de la dieta en la que se redujo el folato, los niveles de HHCY fueron más elevados en los ratones carentes de la enzima CBS.
Modificación de la hiperhomocisteinemia en animales heterocigotos para cistationina betasintasa y metilentetrahidrofolato reductasa mediante el uso de diferentes dietas
| Sustrato | Genotipo | HCY plasmática (μmol/l) | Principales cambios en la dieta | Metionina (g/kg) | Folato (mg/kg) | Referencias |
| C57BL/6J | CBS | 140±51 | Muy alta en metionina y baja en folato | 20 | 3 | 230 |
| C57BL/6J | MTHFR | 9±3 | 51 | |||
| C57BL/6J | CBS | 63±27 | Alta en metionina y restringida en folato | 8 | 0,2 | 49,227,228 |
| BALB/c | MTHFR | 18±3 | 50 | |||
| C57BL/6J | CBS | 37±11 | Estándar, con 5g/l de metionina en agua | 4 | 7±1 | 47,51,213,227,236 |
| BALB/c | MTHFR | 7±1 | 50 | |||
| C57BL/6J | MTHFR | 39±2 | Con folato restringido | 4 | 0,3 | 217 |
| BALB/c | MTHFR | 27±15 | 4 | 0,2 | 50,217 | |
| C57BL/6J | CBS | 9±1 | 5 | 1 | 207,208,213 | |
| C57BL/6J | MTHFR | 23±1 | Baja en folato | 4 | 2 | 217 |
| BALB/c | MTHFR | 12±1 | 217 | |||
| C57BL/6J | CBS | 10±3 | Estándar | 4 | 7±1 | 47,49,51,142,208,22 |
| BALB/c | MTHFR | 5±1 | 50 |
CBS: cistationina betasintasa; HCY: homocisteína; MTHFR: metilentetrahidrofolato reductasa.
Un modelo genético de hipohomocisteinemia se encontró al inactivar el gen de la fosfatidil etanolamina N-metiltransferasa ya que, al no metilarse este fosfolípido, no se emplea la S-adenosil metionina y no se genera en el hígado la suficiente S-adenosil HCY para producir HCY. Los animales carentes de la enzima presentan esteatosis hepática210. Esto puede deberse a que existe una disminución de la actividad lecitina: colesterol aciltransferasa, lo que puede causar una maduración defectuosa de las lipoproteínas de alta densidad (HDL), al igual que la disminución de la betaoxidación de ácidos grasos está disminuida por defecto de la tiolasa211.
La sobreexpresión de la proteína Dyrk-1a aumenta la actividad de la S-adenosil HCY hidrolasa y disminuye los niveles de HCY212. A su vez, la HCY regula los niveles de la proteína Dyrk-1a a través de la acción de la calpaína213. Otro modelo de hipohomocisteinemia se presenta en los ratones carentes del PGC-1α214. Todos estos mecanismos de control apuntan hacia nuevas vías de regulación de la HCY.
Modelos combinados de hiperhomocisteinemia y otros factores de riesgoLa disponibilidad de estos modelos modificados genéticamente abre un nuevo abanico de posibilidades para explorar la combinación de factores de riesgo al objeto de determinar la participación de la HHCY en otros entornos fenotípicos.
El grupo de Wang et al generó ratones dobles deficientes para los genes de CBS y de ApoE. Al analizar su fenotipo, encontraron una arteriosclerosis más pronunciada que la encontrada en los ratones únicamente deficientes para el gen de la ApoE215. También observaron que la existencia de HHCY inducía un aumento en la captación de las LDL acetiladas por los macrófagos y una disminución de las HDL, que podrían estar facilitando el mayor acúmulo de colesterol y triglicéridos en la pared del vaso216. En el caso del doble mutante heterocigoto para MTHFR y homocigoto para ApoE, la presencia de la deficiencia de MTHFR aumentó el acúmulo de lípidos y la hipertrigliceridemia se asoció con los niveles de HCY plasmática217.
Se han generado ratones carentes de CBS que sobreexpresan la proteína humana normal bajo el control de un promotor inducible por zinc. En presencia de zinc, aumenta la actividad de la enzima de dos a cuatro veces con el consiguiente descenso de la HCY en el plasma. De esta manera, consiguen un buen mecanismo para rescatar a los neonatos carentes de CBS que sufren una alta letalidad183. Este mismo grupo ha generado dos nuevos ratones transgénicos que expresan las variantes I278T y T424N de la CBS humana; en estos casos, se observó de nuevo un descenso notable en la mortalidad de los homocigotos pero sorprendentemente no se observa una disminución concomitante de los niveles de HCY. Como hipótesis, estos autores proponen que la enzima CBS puede tener otra función distinta a la que conocemos dentro del catabolismo de la HCY218. Más recientemente, otro grupo ha generado ratones carentes de CBS que sobreexpresan la proteína humana normal o la variante I278T, dos modelos animales con niveles séricos de HCY de 169 y de 296μmol/l, respectivamente. Solo el último grupo de ratones presentó alopecia, osteoporosis, estrés del retículo en el hígado y el riñón, reducción de la supervivencia, elevación de HCY oxidada plasmática y aumento de S-adenosil HCY hepática. Estos modelos sugieren la existencia de un umbral de HCY para producir el efecto patológico219. Con un desarrollo similar de nuevos modelos, en este caso con ratones carentes de CBS que sobreexpresan la variante de proteína humana S466L, se demostró in vivo que esta forma enzimática era inestable y su actividad enzimática era ineficiente, lo que originó homocistinuria220.
La sobreexpresión de la GPx-1 en animales heterocigotos carentes de una copia del gen CBS recuperaba la disfunción endotelial27. Este resultado apunta inequívocamente al papel de los peróxidos en el desarrollo de la disfunción endotelial ocasionada por la HHCY.
Nuestro grupo ha investigado la combinación de la HHCY junto con la hipoalfalipoproteinemia moderada mediante la generación del ratón carente de los genes CBS y ApoA-1. Nuestros resultados muestran que la presencia simultánea de HCY elevada y de niveles bajos de colesterol HDL es un factor temprano de desarrollo de hipertensión arterial al inducir hipertrofia ventricular izquierda y que en el proceso está implicada la menor producción de NO221. Igualmente, la presentación fenotípica es muy variable dependiendo de la raza de ratones empleada, ya que depende también de la expresión de los genes codificantes para proteínas contráctiles222. Utilizando este mismo modelo, hemos demostrado que la simvastatina puede controlar esta hipertensión al restaurar la producción de NO223,224.
ConclusiónLa HCY es un aminoácido cuya elevación sanguínea aparece implicada en múltiples patologías. Sus niveles muestran una enorme variabilidad en función de la raza, el sexo, la edad y otros factores ambientales. En este sentido, el tipo de dieta puede ejercer un papel muy importante y es un factor poco abordado.
El empleo del ratón como modelo experimental en este campo está permitiendo que el progreso de las dietas que influyen en estas patologías haya sido notable. Esto, unido al desarrollo de animales modificados genéticamente con HHCY, está permitiendo caracterizar los mecanismos moleculares implicados en la acción in vivo de la HCY. Además, la combinación de estos modelos con otros modificados genéticamente permite definir la influencia de la combinación de factores de riesgo en el desarrollo de diversas patologías y contribuir así a explicar muchas de las evidencias epidemiológicas en humanos.
FinanciaciónEl trabajo llevado a cabo por este equipo está financiado, en parte, por los proyectos CIBER Fisiopatologia de la Obesidad y Nutrición, una iniciativa del Instituto de Salud Carlos III, CICYT-FEDER (SAF2007-60173), Gobierno de Aragón (PI025/08) y Redes DGA (B-69). M.A.N. y R.C. han sido financiados por un contrato Miguel Servet y por una beca del Gobierno de Aragón, respectivamente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.