





Introducción
El síndrome de Zinsser-Cole-Engman es una enfermedad rara debida a un defecto en la síntesis de la disqueratina humana. Se caracteriza por tener una herencia ligada al cromosoma X, y el gen afectado es el DKC-1. Las manifestaciones cutaneomucosas y hematológicas son las más frecuentes; en el 5% de los pacientes aparecen neoplasias digestivas en el curso evolutivo de la enfermedad.
Caso clínico
Varón de 37 años de edad con antecedentes personales de alergia a la penicilina, bronquitis y enfisema pulmonar (fumador), esquizofrenia paranoide en tratamiento con antipsicóticos, resección de carcinoma epidermoide escrotal con adenopatías inguinales metastásicas, leucoplasia lingual (hiperplasia epitelial benigna) y controlado en dermatología por un síndrome de Zinsser-Cole-Engman (los estudios genéticos confirman la mutación en el gen DKC-1 [5c-T]: sustitución Ala 2 Val), que consultó por cuadro de disfagia para sólidos y vómitos de 10 días de evolución.
Por sus antecedentes personales se le solicitó exploraciones complementarias para el cribado de neoplasias digestivas, y se halló un retraso del vaciamiento gástrico en el tránsito intestinal. La gastroscopia identificó anillos y una membrana esofágica superior, con una úlcera de 1,5 cm de diámetro en incisura angularis, de bordes duros e irregulares y de aspecto maligno. La biopsia informó de adenocarcinoma moderadamente diferenciado de tipo intestinal. Tras el estudio de extensión la estadificación preoperatoria fue cT2N0M0.
Se intervino quirúrgicamente al paciente; se realizó una gastrectomía total con esofagoyeyunostomía en Y de Roux y linfadenectomía D2 (qT2N1M0). La evolución postoperatoria fue satisfactoria. La estadificación definitiva tras el estudio anatomopatológico de la pieza quirúrgica fue de adenocarcinoma moderadamente diferenciado tipo intestinal pT2N1M0.
El tratamiento adyuvante consistió en radioterapia (45 Gy durante 5 sesiones) y quimioterapia con cisplatino.
Discusión y comentarios
El síndrome de Zinsser-Cole-Engman, de baja incidencia, está producido por un defecto en la síntesis de la disqueratina humana, que interviene en la reparación del ADN, la división celular y la síntesis proteica1. Por ello, los órganos que poseen una tasa de reparación y proliferación celular elevada, como la médula ósea, las mucosas y la piel, son los más afectados. Se caracteriza por tener una herencia ligada al cromosoma X en la mayoría de los casos; por tanto, predomina en el sexo masculino2.
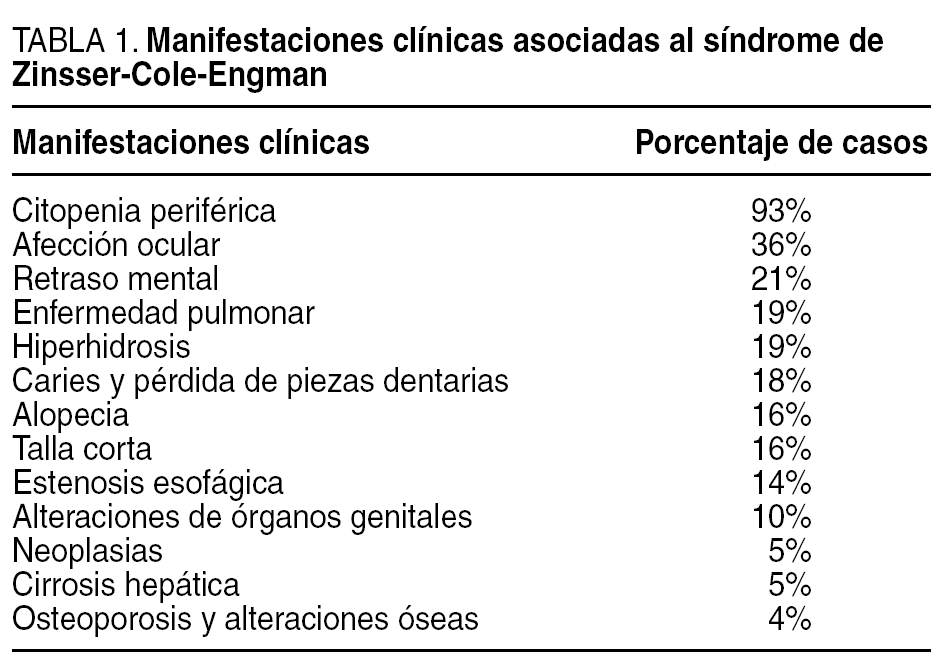
Las manifestaciones cutáneas (distrofia ungueal, alteraciones de la pigmentación cutánea y leucoqueratosis en mucosas) son las primeras en aparecer3, y se diagnostican en etapas tempranas de la vida. En torno a la segunda década, aparecen complicaciones hematológicas (displasia tímica, anemia aplásica y citopenia periférica). Existen otras manifestaciones clínicas debidas al estado de inmunodepresión y a las alteraciones genéticas (tabla 1), entre las que destaca la infección por Pneumocystis carinii4,7. Se ha descrito que las alteraciones oculares, tales como la atresia de los conductos lacrimales, la panuveítis, el síndrome seco, las cataratas y la perforación corneal, acompañan a este síndrome5. Alteraciones esofágicas, como estenosis o membranas, aparecen en aproximadamente un 10-15% de los pacientes6. Un porcentaje menor, en torno al 5%, desarrolla una neoplasia gastrointestinal en el curso de la enfermedad, y hay documentación de neoplasias de recto, canal anal y adenocarcinomas de páncreas, estómago y esófago7,8.
En el paciente que presentamos, las lesiones dérmicas diagnosticadas fueron la leucoplasia oral y el carcinoma epidermoide de escroto. Endoscópicamente se diagnosticó una membrana superior y anillos esofágicos junto a un adenocarcinoma gástrico en incisura angularis. No aparecieron otras complicaciones infecciosas, inmunológicas ni hematológicas.
El diagnóstico se basa en las manifestaciones clínicas típicas, la asociación familiar y el estudio genético, confirmado con la secuenciación del gen DKC-1.
Las causas más frecuentes de muerte en estos pacientes son los fracasos medulares, las alteraciones respiratorias y las neoplasias1.
El único tratamiento capaz de alterar el curso de la enfermedad es el trasplante de médula ósea1. Es obligado el control y el seguimiento de estos pacientes para el diagnóstico precoz de complicaciones infecciosas y neoplásicas, así como el estudio de los familiares más cercanos.
Aunque se trataba de un paciente joven con síntomas digestivos poco específicos de corta evolución, por sus antecedentes de disqueratosis congénita se decidió realizar exploraciones complementarias en busca de neoplasias digestivas, y se diagnosticó de un adenocarcinoma gástrico.
En cuanto al tratamiento del adenocarcinoma gástrico, en este caso se realizó gastrectomía total con esofagoyeyunostomía en Y de Roux, asociado a linfadenectomía D2.
Actualmente, la extensión de la linfadenectomía en la cirugía del cáncer gástrico está en discusión en Occidente por el incremento de la morbimortalidad postoperatoria que supone la resección extensa y la dudosa mejoría en la supervivencia de algunos pacientes9. Los objetivos de la linfadenectomía son minimizar el error en la estadificación, reducir los riesgos de recurrencia locorregional e incrementar el número de pacientes con resección R0. Existe evidencia grado B de que en los casos de cánceres gástricos curables se debe realizar linfadenectomía D210. En cualquier caso, la estrategia terapéutica y el pronóstico dependen, en gran medida, del estadio tumoral y la experiencia del equipo quirúrgico.
El interés de este caso radica en destacar que en los pacientes que presentan una disqueratosis congénita la posibilidad de aparición de una neoplasia digestiva es más elevada que en la población normal y que se pueden beneficiar de su detección precoz, cuando el tratamiento quirúrgico mejora el pronóstico de estas neoplasias. Aunque se trata de un síndrome de baja incidencia, creemos que se debería establecer protocolos sistematizados de exploraciones complementarias para la detección precoz de estos tumores, así como alentar el estudio diagnóstico de este síndrome en familiares de pacientes afectados.
Correspondencia: Dr. M. Bruna Esteban.
Lepanto, 2; bloque 3, pta. 3. 46137 Playa de La Pobla de Farnals. Valencia. España.
Correo electrónico: drbruna@comv.es
Manuscrito recibido el 12-4-2005 y aceptado el 15-7-2005.

