






La muerte súbita cardíaca (MSC) de origen no isquémico es una entidad devastadora que con frecuencia acontece en sujetos jóvenes, aparentemente sanos y de forma inesperada. Su etiología suele ser genética, causada predominantemente por miocardiopatías y canalopatías, que son trastornos monogénicos, es decir, se transmiten a la descendencia por mutaciones en un único gen1. Actualmente disponemos de terapias con eficacia probada para prevenir la MSC, siendo el paradigma el desfibrilador automático implantable (DAI). Para que cumplan su función preventiva deben aplicarse «en tiempo», y por ello resulta imprescindible la detección precoz de los casos subclínicos mediante un estudio; es aquí donde la genética tiene su papel fundamental. En el presente artículo hacemos una revisión del estado actual de la genética en el manejo de las arritmias malignas y la MSC de origen no isquémico.
¿Cuál es la utilidad real y rentabilidad de los tests genéticos en las diferentes enfermedades?La gran utilidad de la genética en el estudio familiar es su alto valor predictivo negativo y positivo en los casos en los que se detecta una mutación claramente asociada con el fenotipo observado, o bien porque esté descrita como asociada de forma consistente en la literatura, o bien por las características moleculares de la mutación. En estos casos, su hallazgo en familiares nos permite realizar un estudio, seguimiento y tratamiento adecuados, y sobre todo su ausencia en otros familiares nos permite descartar en ellos la enfermedad en cuestión, evitando años de incertidumbre y la repetición de pruebas diagnósticas. Sin embargo, el hallazgo de una mutación causal en el estudio genético dirigido no es igual de frecuente en unas enfermedades que en otras.
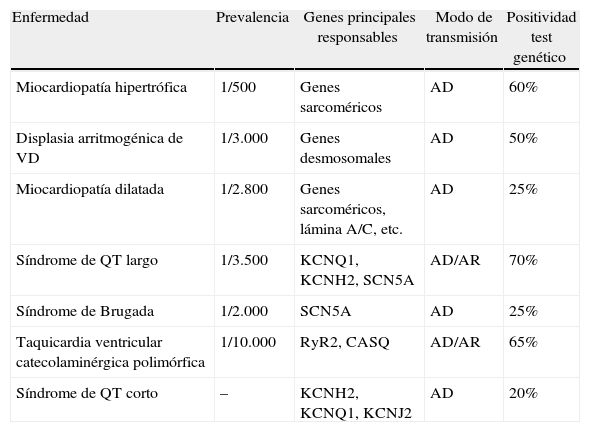
La tabla 1 muestra la base genética y la rentabilidad esperada del test genético en las principales enfermedades hereditarias causantes de MSC. Entre las miocardiopatías, si bien es cierto que hasta en el 30 al 50% de casos de miocardiopatía dilatada se puede demostrar historia familiar2, la rentabilidad del test genético en esta enfermedad es menor que en la miocardiopatía hipertrófica y arritmogénica, donde supera el 50%, concentrado en genes sarcoméricos y desmosomales, respectivamente. Entre las canalopatías las diferencias son, si cabe, más evidentes, con una alta sensibilidad del test genético en el síndrome de QT largo y la taquicardia ventricular catecolaminérgica polimórfica (TVCP) y baja sensibilidad en el síndrome de Brugada y de QT corto. En los 2primeros el hallazgo de mutaciones patogénicas puede llegar al 70-80% según las series, con la mayoría de las variantes localizadas en 2genes para la TVCP y en 3 para el síndrome de QT largo (tabla 1)3.
Bases genéticas de las cardiopatías hereditarias predisponentes a la muerte súbita cardíaca
| Enfermedad | Prevalencia | Genes principales responsables | Modo de transmisión | Positividad test genético |
| Miocardiopatía hipertrófica | 1/500 | Genes sarcoméricos | AD | 60% |
| Displasia arritmogénica de VD | 1/3.000 | Genes desmosomales | AD | 50% |
| Miocardiopatía dilatada | 1/2.800 | Genes sarcoméricos, lámina A/C, etc. | AD | 25% |
| Síndrome de QT largo | 1/3.500 | KCNQ1, KCNH2, SCN5A | AD/AR | 70% |
| Síndrome de Brugada | 1/2.000 | SCN5A | AD | 25% |
| Taquicardia ventricular catecolaminérgica polimórfica | 1/10.000 | RyR2, CASQ | AD/AR | 65% |
| Síndrome de QT corto | – | KCNH2, KCNQ1, KCNJ2 | AD | 20% |
Para el diagnóstico genético de las cardiopatías hereditarias con riesgo arrítmico es esencial disponer de experiencia clínica y asesoramiento genético especializado, ya que con frecuencia se detectan variantes de significado clínico incierto o de dudosa patogenicidad. Esto conlleva el riesgo de catalogar sujetos portadores de variantes no patogénicas como portadores de mutaciones causales de enfermedad. Este hecho, denominado en algunos trabajos como «ruido genético»4, puede orientar el diagnóstico clínico en una dirección equivocada, promover la adopción de medidas terapéuticas erróneas como la retirada del deporte de competición o, en casos extremos, condicionar la inserción de un DAI en un sujeto completamente sano. Además, la detección de mutaciones «privadas», no previamente descritas, es muy frecuente, lo que añade otro punto de complejidad a la interpretación del estudio genético.
Para reducir al máximo la posibilidad de error en un caso concreto, una vez identificada una variante en un gen de dudosa patogenicidad, lo correcto es secuenciarla en el resto de familiares e intentar desenmascarar el fenotipo sugerido por la variante en los portadores; por ejemplo, si identificamos una variante de significado incierto en KCNQ1 en un sujeto con intervalo QTc en límite superior de la normalidad estudiado por un antecedente familiar de muerte súbita, antes de atribuir el fenotipo familiar a esta variante se debería poder demostrar el fenotipo de QT prolongado en los portadores, mediante ECG basal, ergometría o test de epinefrina.
El hecho contrario también sucede: no encontrar una variante genética en los genes secuenciados en un paciente con un fenotipo evidente no supone en ningún caso ausencia de enfermedad, y el resultado debe interpretarse como un falso negativo de la genética.
Para evitar errores, cada vez que se identifique una variante genética en un sujeto debemos responder a las siguientes preguntas:
- -
¿Ha sido previamente descrita en algún artículo o comunicación como mutación causal de la enfermedad sospechada? ¿Con qué grado de evidencia clínica? ¿Existía co-segregación del fenotipo con el genotipo en las familias portadoras?
- -
¿Qué características moleculares tiene la variante? ¿Afecta a una región determinante de la proteína, o por el contrario es una zona intrónica o con poca repercusión funcional? ¿Qué probabilidad de patogenicidad le confieren los sistemas bioinformáticos de predicción? ¿Afecta a un residuo conservado entre especies?
- -
¿Se ha detectado la variante previamente en controles sanos? ¿Con qué frecuencia?
Responder a estas preguntas no es sencillo. El hecho de contar con profesionales con experiencia en genética cardiovascular, con bases de datos amplias de mutaciones publicadas y «privadas», así como la posibilidad de realizar estudios funcionales en modelos celulares/animales, minimiza la posibilidad de errores de interpretación y supone, a día de hoy, un componente imprescindible para desarrollar una consulta de cardiopatías familiares.
¿Qué actitud tomar con un portador silente? Importancia de la penetrancia clínica incompletaUno de los problemas más frecuentes en la práctica clínica de estas enfermedades, sobre todo en el caso de las canalopatías, es la ausencia de una clara manifestación fenotípica en familiares portadores de la mutación «patogénica» causante de la enfermedad en el probando. Incluso en ocasiones es el propio caso índice el que no muestra datos fenotípicos evidentes aun a pesar de haber experimentado episodios clínicos graves como síncope o paro cardíaco, siendo los test genéticos la única vía para llegar al diagnóstico en estos raros casos.
Las canalopatías, y en menor medida las miocardiopatías, muestran una penetrancia clínica incompleta que explica que los portadores de la mutación causal presenten una expresión fenotípica negativa o solo parcial de la enfermedad respecto a la que muestra el probando con igual mutación. En algunas ocasiones de fenotipo aparentemente negativo el uso de test de provocación farmacológica con epinefrina (QT largo) y flecainida (síndrome de Brugada), o de técnicas de imagen más sensibles como la cardiorresonancia (miocardiopatías), puede desenmascarar el fenotipo latente. Esto se traduce en que una gran proporción de familiares asintomáticos portadores de mutación no cumplen los criterios diagnósticos vigentes para la enfermedad concreta5. El caso mostrado en la figura 1 es un ejemplo típico de nuestra actividad cotidiana en una consulta de cardiopatías familiares, donde muchos miembros de una misma familia, portadores de una misma variante patogénica en KCNH2 para síndrome de QT largo muestran diferentes intervalos QTc, algunos de ellos en rango normal. Ello resalta el valor del test genético en la identificación de estos familiares, y posteriormente, para desenmascarar el patrón fenotípico, la realización de pruebas más específicas en estos portadores silentes, que en el caso mostrado serían la ergometría y/o test de epinefrina.

Baja penetrancia en un caso de síndrome de QT largo familiar con mutación en KCNH2. Nótese la diferente expresividad clínica en el ECG, mayor en los miembros de la tercera generación.MSC: muerte súbita cardíaca; los círculos indican mujeres; los cuadrados, varones; en negro, los portadores de la variante.
Así como la evidencia a favor del uso de los tests genéticos en el diagnóstico de probando y familiares está suficientemente validada, como se ha descrito previamente, no sucede lo mismo respecto a su capacidad para apoyar la estratificación pronóstica y la toma de decisiones clínicas. Existen algunas enfermedades en las que el hallazgo de mutaciones en determinados genes conlleva ciertos rasgos fenotípicos concretos, como en el caso de la miocardiopatía hipertrófica y el gen de la troponina T, cuyas mutaciones provocan fenotipos de hipertrofia ligera con incidencia algo más elevada que otros loci genéticos de muerte súbita. Otro ejemplo es el síndrome de QT largo, donde las mutaciones en los 3genes principales se traducen en un fenotipo electrocardiográfico concreto para cada gen y en una respuesta diferente al tratamiento con betabloqueantes. Respecto a la capacidad puramente pronóstica de MSC, la información es mucho más confusa y contradictoria. Por ejemplo, se ha descrito en el síndrome de Brugada que ser portador mutacional en SCN5A conlleva mayor riesgo de MSC, pero esto no ha sido confirmado en los grandes registros publicados6. En el síndrome de QT corto los escasos trabajos relevantes disponibles parecen mostrar una mayor respuesta al tratamiento con quinidina en los sujetos portadores de variantes en el gen HERG (KCNH2), aunque este hecho se concluye de estudios con escaso tamaño muestral. Por todo ello, nunca debe tomarse aisladamente la información genética para estimar el riesgo de padecer MSC y, por supuesto, para decidir el implante de un DAI.
En la estratificación del riesgo de muerte súbita resulta crucial individualizar los tratamientos en cada caso, basándose fundamentalmente en los episodios y en el fenotipo clínico del sujeto y de la familia. Los datos genéticos tendrán un papel secundario de apoyo a la información clínica.
¿Qué papel tienen los tests genéticos en el abordaje diagnóstico de la muerte súbita cardíaca de origen incierto?Existe una pequeña proporción de casos de MSC en los que, a pesar de los estudios convencionales, no se consigue llegar al diagnóstico final. En el caso de que el paciente haya sobrevivido al paro cardíaco el episodio se catalogaría de fibrilación ventricular idiopática, y la probabilidad de no encontrar una etiología es inferior a los casos en que el probando fallece.
La figura 2 muestra el algoritmo que proponemos para el estudio de la MSC, con un papel concreto para el uso de los tests genéticos. Tanto en el caso de superviviente como del fallecido por MSC, antes de indicar un estudio genético es primordial obtener toda la información clínica posible que permita guiar y orientar el resultado de un test genético. Esto es más complejo en caso del sujeto fallecido, ya que en muchas ocasiones no habrá sido estudiado previamente, y la autopsia muchas veces, o no se realiza, o aporta información limitada por la escasa disponibilidad de forenses con formación específica en este tipo de patologías. En ambos casos resulta de gran utilidad la realización de estudio familiar con pruebas básicas, como electrocardiograma y ecocardiograma, ya que en ocasiones el probando no muestra datos fenotípicos y es un familiar el que aporta la «pista» para el diagnóstico definitivo. En el caso de hallar algún indicio fenotípico en cualquiera de estos pasos diagnósticos, el test genético estaría indicado con la técnica convencional de Sanger, siendo de gran utilidad para los familiares en el caso de hallar una mutación patogénica concordante con el fenotipo observado.
.")
En los casos en los que no hay ningún dato fenotípico para sospechar la etiología de la MSC, el test genético debe ser interpretado con precaución. Actualmente disponemos de tecnología sofisticada con el método Next Generation Sequencing, que permite secuenciar gran cantidad de genes de forma fiable y a un costo similar a la tecnología convencional por Sanger. De esta forma, en estos casos de fenotipos poco aparentes o mixtos puede ser de gran utilidad el hecho de hallar una variante patogénica con evidencia de causalidad sólida. Sin embargo, el hecho de tener la posibilidad de secuenciar numerosos genes incrementa el riesgo de falsos positivos, y conviene ser muy cuidadoso a la hora de interpretar los hallazgos. Este es el motivo por el que en las últimas recomendaciones de la Heart Rhytm Society y de la European Heart Rythm Association, en los casos sin fenotipo asociado el test genético se cataloga de indicación claseiii7. En nuestra experiencia, la autopsia molecular (estudio genético sobre muestras del sujeto fallecido) o el test genético en el superviviente a un paro cardíaco de origen incierto tienen su utilidad en el caso de hallar una variante patogénica, en general de una canalopatía subclínica, basándonos en la más que demostrada baja penetrancia en el fenotipo de estas enfermedades.
ConclusionesEl estudio genético tiene un papel fundamental en el estudio familiar de miocardiopatías y canalopatías. Permite, por un lado, la identificación de portadores silentes, que pueden beneficiarse de una terapia eficaz y modificación de estilo de vida, y por otro, evita la incertidumbre y la repetición de pruebas seriadas en los no portadores. La utilidad diagnóstica en el probando es más limitada, siendo rentable sobre todo en los casos con fenotipo incierto o borderline, y en cualquier caso sin valor pronóstico, pues una vez que se ha identificado la enfermedad el manejo clínico no diferirá en función del genotipo. Y es que a día de hoy es limitada la utilidad pronóstica, y debe evitarse la toma de decisiones terapéuticas basadas en el hallazgo de «mutaciones de alto riesgo» sin considerar los datos clínicos (fig. 3). Es crucial la colaboración entre clínicos y genetistas para la correcta interpretación de los hallazgos genéticos como forma de evitar los falsos positivos, especialmente cuando no exista un fenotipo claro que sugiera la etiología del episodio de MSC.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.

