




Como se ha planteado en anteriores artículos de esta serie, en los últimos años las enfermedades raras (ER) son objeto de interés para todos los estamentos del Sistema Nacional de Salud. Por este motivo, los médicos de atención primaria no pueden quedarse al margen y han de comenzar a colaborar de forma activa en la detección precoz de pacientes con ER —a fin de acelerar su acceso a un diagnóstico preciso— y realizar un seguimiento adecuado de estos pacientes y sus familias. El objetivo de estos artículos es informar de forma breve, clara y concisa del procedimiento adecuado para lograr esta meta en algunas de las ER más frecuentes en nuestro país. Como hemos comentado, el número de ER es muy elevado, por lo que para la selección de las afecciones el Grupo de Trabajo de la Sociedad Española de Medicina Familiar y Comunitaria (semFYC) sobre Genética Clínica y ER ha decidido comenzar con aquellas cuyas asociaciones de pacientes han mantenido una actitud más «proactiva» con el Grupo de Trabajo, entendiendo que esto implica mayor necesidad de información al respecto. Por ello, desde estas líneas, animamos a profesionales y asociaciones a ponerse en contacto con el Grupo de Trabajo para solicitar información sobre otras ER que quieran ver reflejadas en artículos posteriores. En esta ocasión nos ocuparemos del síndrome de Marfan.
El síndrome de Marfan es una enfermedad hereditaria rara, debida a un trastorno genético de herencia autosómica dominante, que debe su nombre al médico francés Jean-Bernard Antoine Marfan, que la describió por primera vez en 1896 en un niño de 5 años con dedos y extremidades más largas de lo normal, acompañado de otras alteraciones esqueléticas. Afecta al tejido conectivo, a sus fibras elásticas, manifestándose especialmente en los sistemas u órganos que mayor cantidad de ellas tienen, tales como el cardiovascular, el esquelético, los ojos, los pulmones, la membrana fibrosa que recubre el cerebro (duramadre) y la espina dorsal (sistema nervioso).
Este tejido conectivo está constituido por una red de microfibrillas, que está formada por fibrilina, codificada en el gen FBN1 en el cromosoma 15q21.
De este gen se han descubierto más de 500 mutaciones, casi todas únicas para un individuo o una familia afectada1. Ocasionalmente también afecta al gen TGFBN2.
Incidencia y prevalenciaLa incidencia ha sido descrita entre 1/3.000 y 1/20.000 habitantes, aunque se acepta actualmente que la incidencia mínima de nacimientos al año se establece en 1/9.800. La prevalencia en estudios americanos y basados en su población se aproxima a 1/5.000 habitantes.
El síndrome de Marfan es el resultado de una alteración de la síntesis de fibrilina-1. La codificación del gen de esta proteína (FBN1) ha sido parcialmente clonada y fue localizada en el cromosoma 15q21 en los humanos en 1991. Un segundo gen implicado en el síndrome de Marfan (denominado síndrome de Marfan 2)2 fue localizado en 1994 tras estudiar a una familia francesa afectada de síndrome de Marfan, de la que se había excluido la implicación del gen FBN1, lo que llevó a pensar en que hubiera heterogeneidad genética. El segundo gen implicado fue localizado en el brazo corto del cromosoma 3, el 3p25. Alteraciones en este gen explican un 8–15% de los casos de síndrome de Marfan.
Criterios de sospecha diagnóstica en atención primariaEl diagnóstico a veces es complicado, ya que está basado en la presencia de varios signos o síntomas en varios sistemas y a menudo requiere la consulta y el consejo de diversos especialistas.
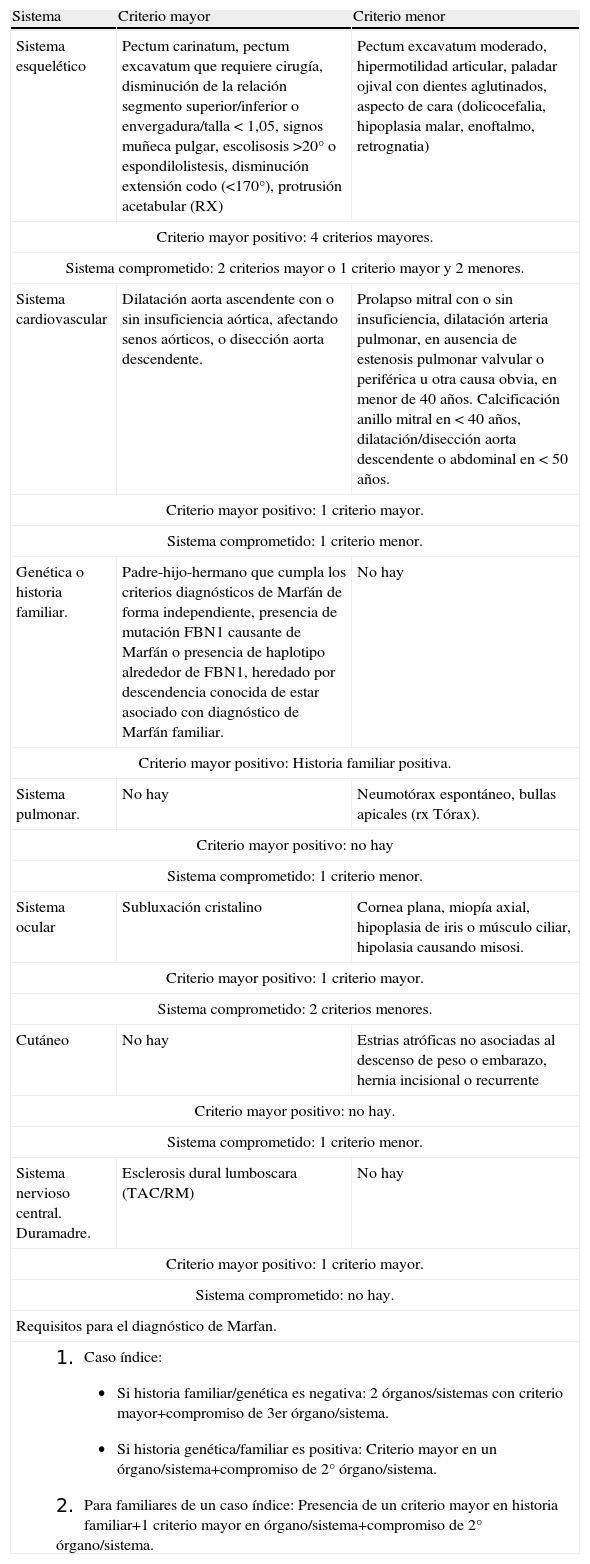
Los criterios diagnósticos del síndrome de Marfan fueron establecidos durante el séptimo Congreso Internacional de Berlín en 1986. Con el tiempo Paepe revisó dichos criterios y creó los criterios nosológicos de Ghent en 1996, los cuales se basan en la historia familiar-genética y la afección de diferentes órganos o sistemas revisados (tabla 1).
Criterios diagnósticos del síndrome de Marfan (NOSOLOGIA GHENT, Paepe et al, 1996)
| Sistema | Criterio mayor | Criterio menor |
| Sistema esquelético | Pectum carinatum, pectum excavatum que requiere cirugía, disminución de la relación segmento superior/inferior o envergadura/talla < 1,05, signos muñeca pulgar, escolisosis >20° o espondilolistesis, disminución extensión codo (<170°), protrusión acetabular (RX) | Pectum excavatum moderado, hipermotilidad articular, paladar ojival con dientes aglutinados, aspecto de cara (dolicocefalia, hipoplasia malar, enoftalmo, retrognatia) |
| Criterio mayor positivo: 4 criterios mayores. | ||
| Sistema comprometido: 2 criterios mayor o 1 criterio mayor y 2 menores. | ||
| Sistema cardiovascular | Dilatación aorta ascendente con o sin insuficiencia aórtica, afectando senos aórticos, o disección aorta descendente. | Prolapso mitral con o sin insuficiencia, dilatación arteria pulmonar, en ausencia de estenosis pulmonar valvular o periférica u otra causa obvia, en menor de 40 años. Calcificación anillo mitral en < 40 años, dilatación/disección aorta descendente o abdominal en < 50 años. |
| Criterio mayor positivo: 1 criterio mayor. | ||
| Sistema comprometido: 1 criterio menor. | ||
| Genética o historia familiar. | Padre-hijo-hermano que cumpla los criterios diagnósticos de Marfán de forma independiente, presencia de mutación FBN1 causante de Marfán o presencia de haplotipo alrededor de FBN1, heredado por descendencia conocida de estar asociado con diagnóstico de Marfán familiar. | No hay |
| Criterio mayor positivo: Historia familiar positiva. | ||
| Sistema pulmonar. | No hay | Neumotórax espontáneo, bullas apicales (rx Tórax). |
| Criterio mayor positivo: no hay | ||
| Sistema comprometido: 1 criterio menor. | ||
| Sistema ocular | Subluxación cristalino | Cornea plana, miopía axial, hipoplasia de iris o músculo ciliar, hipolasia causando misosi. |
| Criterio mayor positivo: 1 criterio mayor. | ||
| Sistema comprometido: 2 criterios menores. | ||
| Cutáneo | No hay | Estrias atróficas no asociadas al descenso de peso o embarazo, hernia incisional o recurrente |
| Criterio mayor positivo: no hay. | ||
| Sistema comprometido: 1 criterio menor. | ||
| Sistema nervioso central. Duramadre. | Esclerosis dural lumboscara (TAC/RM) | No hay |
| Criterio mayor positivo: 1 criterio mayor. | ||
| Sistema comprometido: no hay. | ||
| Requisitos para el diagnóstico de Marfan. | ||
| ||
El síndrome de Marfan se caracteriza por la aparición de una serie de datos físicos característicos que afectan a diversos órganos o sistemas.
- 1.
Afección cardiovascular:
- •
Prolapso de la válvula mitral y regurgitación. Posiblemente cause la insuficiencia mitral, que supone una complicación severa en pacientes jóvenes.
- •
Dilatación ventricular izquierda.
- •
Dilatación de arteria pulmonar.
- •
Dilatación de la raíz de la aorta, que adquiere la forma típica de una cebolla. Esta dilatación se asocia normalmente a una incompetencia de la válvula aórtica. Constituye la principal causa de muerte, aunque ha aumentado la esperanza de vida de los pacientes con los tratamientos aplicados (de una media de edad al fallecimiento de 32±16 años en 1972 a 45±17 años en 1998).
- •
- 2.
Afectación ocular:
- •
Bilateral, subluxación del cristalino (40–56%).
- •
Miopía (28%).
- •
Desprendimiento de retina (0,78%)3.
- •
- 3.
Afección del aparato locomotor:
- •
Excesivo crecimiento de las extremidades (dolicoestenomelia), es la anormalidad fenotípica fundamental, que se acentúa porque no hay aumento paralelo de la grasa ni de la masa muscular.
- •
La apariencia de los afectados de síndrome de Marfan es muy caracterísitica: muy alto, gran envergadura, muy delgado, deformidad de tronco, y dedos desproporcionadamente largos y delgados (aracnodactilia), reconocible mediante el signo de Walker-Murdoch, en el que, debido a estos dedos excesivasmente largos y la delgadez del antebrazo, el dedo pulgar y el meñique se cruzan al abarcar la muñeca contralateral4.
- •
Disminución mineral ósea en columna vertebral y cadera, pero no se ha observado aumento de fracturas.
- •
La hipermotilidad articular es muy corriente y afecta al 85% de los pacientes menores de 18 años y el 56% de los adultos; estos pacientes sufren artralgias, mialgias y lesiones ligamentosas.
- •
Afección de la columna, con escoliosis muy marcada, que afecta al 60% de los pacientes.
- •
Protrusión acetabular en el 40% de los pacientes.
- •
Afección de caja torácica, con aparición de deformidad hacia fuera (pectus carinatum) o hacia dentro (pectus excavatum).
- •
- 4.
Afección del aparato respiratorio:
- •
Puede aparecer un patrón ventilatorio restrictivo en pacientes con pectus excavatum severo que ocurre en 2/3 de los pacientes con síndrome de Marfan.
- •
Un 4–11% de los pacientes pueden sufrir neumotórax espontáneo, que puede estar asociado a ampollas apicales.
- •
Pacientes adultos con síndrome de Marfan tienen una tendencia aumentada al colapso de las vías aéreas durante el sueño, lo que causa las conocidas apneas obstructivas del sueño, que pueden favorecer la somnolencia diurna.
- •
- 5.
Afección de la boca:
- •
Paladar arqueado; la mayoría de las veces faltan los pilares del velo del paladar y hay apiñamiento de los dientes que produce mala oclusión.
- •
- 6.
Afección del sistema nervioso central:
- 7.
Síndrome de Marfan y embarazo:
- •
En el embarazo el riesgo de disección aórtica, aproximadamente un 4,5% de los embarazos, está aumentado debido a la inhibición del colágeno y la elastina por estrógenos y el estado circulatorio de hipervolemia.
- •
Se debe asesorar a los pacientes con síndrome de Marfan desde atención primaria mediante un consejo genético si desean tener descendencia.
Para tratar la afección cardiovascular, se debe plantear el uso de bloqueadores beta, incluidos los niños. Si no toleran estos fármacos, se podría utilizar antagonistas del calcio o inhibidores de la enzima de conversión de angiotensina (IECA).
Si falla el tratamiento farmacológico y la raíz aórtica es >5cm, se debería considerar la profilaxis con cirugía.
En la afección ocular la valoración oftalmológica es importante y la valoración regular de la agudeza visual está particularmente recomendada en la infancia. La profilaxis con láser realizando vitrectomía para prevenir el desprendimiento de retina puede ser efectiva en algunos pacientes.
A pesar de la morbilidad y la mortalidad del síndrome de Marfan, un apropiado manejo médico y quirúrgico puede mejorar y alargar la vida de muchos pacientes, y debemos continuar en la búsqueda de nuevos remedios para el futuro7.
Enlaces de interés relacionados con la enfermedadAsociación de afectados del síndrome de Marfan en España (SIMA). San Agatángelo, 44, bajo izquierda. 30007 Alicante. Tel.: 966141580, 619191665. Correo electrónico: sima@marfansima.org. Web:http://www.marfansima.org/.
Asociación española para el Registro y Estudio de las malformaciones Congénitas (ASEREMAC). Facultad de Medicina. Universidad Complutense. 28040 Madrid. Tel.: 913941587, 913941591.
Asociación para las Deficiencias que afectan al Crecimiento y al Desarrollo (ADAC). Enrique Marco Dorta, 6. 41018 Sevilla. Tel.: 902195246, 954989889. Correo electrónico: a.d.a.c@telefonica.net. Web:http://www.geocities.com/HotSprings/Villa/4521/.
Fundación 1000 para la Investigación de los Defectos Congénitos. Serrano, 140. 28006 Madrid. Tel.: 913941587. Web:http://www.fundacion1000.es/.
Federación Española de Asociaciones de Enfermedades Raras (FEDER). Enrique Marco Dorta, 6 local. 41018 Sevilla. Tel.: 954989892. Correo electrónico: f.e.d.e.r@teleline.es. Información y contacto: 902181725; info@enfermedades-raras.org. Web:http://www.enermedades-raras.org/es/default.htm.
- •
El síndrome de Marfan es una enfermedad hereditaria, rara, de herencia autosómica dominante, que afecta al tejido conectivo, a sus fibras elásticas y los sistemas con mayor proporción en este tipo de tejidos.
- •
No tiene tratamiento curativo en conjunto. El tratamiento será sintomático.
- •
Diagnóstico complicado. Nos basaremos en los criterios nosológicos de Ghent.
- •
El objetivo desde atención primaria es asesorar a las parejas afectadas para que consigan consejo genético si desean descendencia y un adecuado manejo médico y quirúrgico de las diversas afecciones para mejorar y alargar la vida de los pacientes.
