







El síndrome de Andersen-Tawil resulta de la alteración de canales de potasio, se hereda de forma autosómica dominante y se cataloga como el tipo 7 de los síndromes de QT largo congénitos. El gen afectado es el KCNJ2, el cual codifica la proteína Kir2.1 que forma el canal de potasio rectificador interno («inward rectifier»). Este canal interviene en la estabilización del potencial de membrana en reposo y controla la duración del potencial de acción en el sistema musculoesquelético y cardíaco. En miocitos ventriculares, es un componente responsable de la rectificación de la corriente de potasio en la fase 3 del potencial de acción. Debido a que Kir2.1 está presente en el sistema musculoesquelético, corazón y cerebro, las alteraciones de esta proteína dan origen a las principales características del síndrome: parálisis flácida, arritmias ventriculares y alteraciones leves a moderadas en el desarrollo del esqueleto, especialmente en manos y pies. En la presente revisión se aborda esta enfermedad desde el punto de vista del diagnóstico clínico y molecular con énfasis en sus manifestaciones cardíacas.
The Andersen-Tawil syndrome is a cardiac ion channel disease that is inherited in an autosomal dominant way and is classified as type 7 of the congenital long QT syndromes. Affected gene is KCNJ2, which forms the inward rectifier potassium channel designated Kir2.1. This protein is involved in stabilizing the resting membrane potential and controls the duration of the action potential in skeletal muscle and heart. It also participates in the terminal repolarization phase of the action potential in ventricular myocytes and is a major component responsible for the correction in the potassium current during phase 3 of the action potential repolarization. Kir 2.1 channel has a predominant role in skeletal muscle, heart and brain. Alterations in this channel produce flaccid paralysis, arrhythmias, impaired skeletal development primarily in extremities and facial area. In this review we address the disease from the point of view of clinical and molecular diagnosis with emphasis on cardiac manifestations.
El síndrome de Andersen-Tawil (ATS, por sus siglas en inglés) es una enfermedad de canales iónicos donde el gen afectado, el KCNJ2 (locus 17q23-q24.2), codifica el canal de potasio rectificador interno Kir2.11. Se cataloga como el tipo 7 de los síndromes de QT largo (SQTL) congénito2, aunque existe cierta controversia al respecto3. En esta revisión se aborda a la enfermedad desde el punto de vista del diagnóstico clínico y molecular con énfasis en sus manifestaciones cardíacas.
AntecedentesEl primer caso clínico de esta enfermedad fue informado por Ellen Andersen, Peter Krasilnikoff y Hans Overvad del Hospital Universitario de Copenhague (Dinamarca), en la revista Acta Pediatrica Scandinavica (1971) y correspondió a un niño de 8 años de edad con 3 características clínicas principales: debilidad muscular (parálisis periódica) intermitente, arritmias (extrasístoles ventriculares) y alteraciones en el desarrollo (talla baja, cabello delgado, dolicocefalia, puente nasal ancho, implantación baja de pabellones auriculares, paladar hendido, dentición retrasada e incompleta, hipoplasia mandibular, pliegue palmar transverso, clinodactilia del quinto dedo de las manos y pies, así como mineralización incompleta del cráneo)4. Tawil et al.5 informaron de 4 pacientes con fenotipo similar e hicieron una mejor descripción del espectro clínico de la enfermedad. Fueron los primeros en utilizar el epónimo síndrome de Andersen. Aunque se señaló en su momento que probablemente el nombre correcto debía ser síndrome de Klein-Lisak-Andersen, debido a la existencia de publicaciones anteriores a Andersen por parte de los 2 primeros autores6, este epónimo no prosperó y tiempo después pasó a denominarse síndrome de Andersen–Tawil debido a que el Dr. Tawil desarrolló los criterios diagnósticos y demostró que este síndrome es distinto de otras formas de parálisis periódica sensible al potasio por medio de un «estudio de ligamiento», señalando incluso la posibilidad de que se tratase de una forma distinta de SQTL7. En 1997, Sansone et al. enfatizaron la presencia de la tríada clásica en 11 pacientes7. El primer informe en México del síndrome fue de Canún, Pérez y Beirana8 y consistió en una descripción clínica muy completa en 3 generaciones de una misma familia. La muestra sanguínea del caso índice y sus familiares fue enviada a EE. UU. donde, junto con pacientes de otras latitudes, se describió por primera vez la mutación causante del síndrome en el gen KCNJ29.
Bases moleculares de la enfermedadMediante un estudio de «análisis de ligamiento» con 400 marcadores polimórficos en el genoma de 15 individuos con ATS, Plaster et al.9 identificaron que la región candidata se localizaba en el locus 17q23 por medio del marcador D17S949 10, obteniendo un lod=3.23 y theta=0.0. Como en este locus se encuentra un gen que codifica para un canal de potasio, el KCNJ2, decidieron estudiarlo, identificando la mutación D71V en la familia mexicana informada previamente por Canún et al.8. Esta mutación tiene un efecto dominante negativo y corresponde a una transversión (cambio de adenina por timina) en el nucleótido 440 que resulta en un cambio en el dominio aminoterminal en la última posición de una alfa-hélice; este residuo es apenas distal al dominio putativo de interacción N-terminal11,12.
Actualmente se conoce que el gen KCNJ2 se localiza en la región 17q23.1-q24.2, tiene una longitud de 11,372 pares de bases, su marco de lectura no se encuentra interrumpido por intrones, tiene 2 exones y 2 transcritos, uno de 5,392 y otro de 1,685 pares de bases. Codifica para una proteína que forma el canal rectificador interno de potasio llamado Kir2.1. Este canal, formado por 427 aminoácidos, tiene un peso molecular de 48kD y 2 regiones membranales conectadas por un dominio en forma de poro y dominios N- y C-terminales citoplasmáticos13,14 (fig. 1).
Kir2.1 es un canal rectificador interno «fuerte», contribuye a la corriente IK1 que interviene en la estabilización del potencial de membrana en reposo y controla la duración del potencial de acción en el sistema musculoesquelético y cardíaco; tiene un papel en la fase de repolarización terminal del potencial de acción. En miocitos ventriculares es un componente mayor responsable de la rectificación en la corriente de potasio en la fase 3 del potencial de acción15,16. La actividad eléctrica espontánea del ventrículo, secundaria a supresión de la corriente de potasio, puede explicar la mayor frecuencia de ectopia ventricular en el ATS a diferencia de otros tipos de SQTL. La pérdida de función de Kir2.1, secundaria a diferentes mutaciones, prolonga el potencial de acción ventricular lo que se traduce en un alargamiento del intervalo QT-U y favorece la aparición de taquiarritmias ventriculares. Las subunidades alfa de los canales Kir están formadas por 2 segmentos transmembranales y un poro. Debido a que estas 4 subunidades alfa están ensambladas para funcionar como un tetrámero, se considera que una mutación en una sola región crítica puede ser suficiente para dañar la función del canal completo11. A la fecha, 2 sitios de mutación reconocidos se encuentran en los residuos de unión a fosfatidilinositol-4-5-bifosfato localizado en el extremo C-terminal y en el asa («loop») del poro o la región externa de Kir2.1. Estas mutaciones causan modificación de la hélice del poro que alteran la selectividad y conformación del filtro o interfieren con el desempeño de la hélice al disminuir la barrera electrostática para la conducción iónica15–18.
Las deleciones de los residuos 95-98 remueven una región del primer dominio transmembranal de Kir2.1. La alteración de estos 4 aminoácidos cambia la formación de la alfa-hélice en la membrana y consecuentemente la inserción del canal en la membrana; adicionalmente no se puede mantener un estado abierto o incluso manejar los iones de potasio19. Andelfinger et al.20 identificaron una mutación de cambio de sentido, arginina 67 por triptófano (R67W), en 41 miembros de una familia en la cual no todos los individuos tenían manifestaciones de ATS, lo que sugiere penetrancia incompleta. En una familia con la mutación R67W en KCNJ2 se observó parálisis periódica solo en hombres, síntomas cardíacos en mujeres y anomalías congénitas en ambos. Este tipo de segregación, limitada al sexo, no ha sido confirmada. Aunque en la actualidad no existe dentro del gen KNCJ2 una mutación predominante o un sitio que concentre la mayoría de las mutaciones («sitio caliente» o «hot-spot» en inglés), cada vez se informan más pacientes con ATS con la mutación R67W, por lo que el sitio donde se localiza esta mutación se ha llegado a considerar como un potencial «hot-spot»21,22.
Hasta la fecha se han descrito 62 mutaciones en el gen KCNJ2 para ATS, la mayoría se relacionan con un efecto dominante negativo (cuando interaccionan 2 o más proteínas, pero una de ellas es anómala [desestabilizando de esta forma el tetrámero]) aunque también hay casos de haploinsuficiencia (cuando disminuye la cantidad de proteína necesaria para formar el canal)11,19–22. También se ha encontrado pérdida de la función del canal debida a una alteración que impide el transporte de las proteínas del canal desde el retículo endoplásmico hacia la membrana23. En la figura 1 se presenta un esquema de la localización de las 62 mutaciones descritas hasta el momento en el canal Kir2.1. Se puede apreciar que la mayoría ocurren en ambas «colas citoplasmáticas», predominantemente en el extremo carboxiterminal.
.")
Mutaciones en el canal Kir2.1. Se puede apreciar que la mayoría ocurren en el dominio C-terminal. Con asterisco, las mutaciones descritas en familias mexicanas. La D71V identificada por Plaster et al.9 y corroborada en el INCICh en los demás miembros de la familia y la R218W identificada por Sistemas Genómicos® S.L. (Valencia España).
Kir2.1 tiene una función predominante en el sistema musculoesquelético, corazón y cerebro, por lo que la alteración de este canal en el ATS conlleva: parálisis flácida, arritmias ventriculares y alteraciones leves a moderadas en el desarrollo del esqueleto24–28. La parálisis flácida por la incapacidad para propagar el potencial de acción en la membrana muscular es resultado de la despolarización sostenida de la membrana celular26,27. La prolongación del intervalo QT-U y las arritmias ventriculares son causadas por la alteración en la repolarización ventricular25,28. Aunque el papel de Kir2.1 en las alteraciones del desarrollo esquelético está por ser aclarado, la corriente de potasio en Kir2.1 es necesaria para la función de los osteoblastos. Se sabe que el intercambio de iones potasio por hidrógeno es necesario para mantener el medio ácido adecuado para una reabsorción ósea normal. Estudios realizados en ratones modificados por ingeniería genética para que uno o más de sus genes estén inactivos muestran letalidad perinatal por deshidratación o insuficiencia respiratoria secundarias a paladar hendido, anomalía que ocurre en el 100% de ellos29. Se desconoce la razón por la cual este modelo no experimenta arritmias ni parálisis periódica.
Para explicar la conocida variabilidad en la expresión fenotípica del ATS se han postulado varias hipótesis. Una de ellas la considera consecuencia de heteromultimerización (unión de subunidades de diferentes proteínas) de Kir2.1 con homólogos de la familia Kir2.x, específicamente Kir2.2 y Kir2.330. Otro gen que interactúa con Kir2.1 es el de la anquirina B, proteína que funciona como un adaptador proteico que está relacionada con el ensamble de la ATPasa de sodio/potasio. Algunas otras proteínas que interactúan con Kir2.1 son la proteína cinasa de anclaje 79, la proteína asociada a sinapsis 97, el complejo de proteína de densidad postsináptica 95/proteína sináptica asociada y la filamina A19.
Características clínicasUno de los primeros síntomas es la debilidad muscular después de una actividad física prolongada; esta comienza en la infancia o adolescencia, puede remitir espontáneamente y su duración va desde unas horas a días. La prueba neurofisiológica (electromiografía pre- y postejercicio) de McManis modificada por Fournier es de utilidad para la detección de ATS en pacientes con parálisis periódica26,27. Las crisis de parálisis periódica no parecen depender de la concentración de potasio, que puede estar alta, normal o disminuida por lo que idealmente la medición de este electrólito debe realizarse durante dichos episodios. En el subgrupo de pacientes donde hay hipocaliemia hay una respuesta satisfactoria a la administración de potasio24. En la tabla 1 se mencionan los criterios diagnósticos para ATS.
Criterios diagnósticos de síndrome de Andersen-Tawil
| Clínicamente sospecha de ATS en individuos con características de la sección A o B: | |
| Sección ADos de 3 criterios | I. Parálisis periódica |
| II. Arritmia sintomática o evidencia electrocardiográfica de ectopia ventricular, aumento en el voltaje de la onda U o de alargamiento del intervalo QTc o Q-Uc | |
| III. Facies característica (frente amplia, fisuras palpebrales cortas, puente nasal amplio, punta de la nariz bulbosa, hipoplasia mandibular y maxilar, labio superior delgado y cara triangular) alteraciones dentales, manos y pies pequeños, y al menos 2 de los siguientes datos:1. Implantación baja de pabellones auriculares2. Hipertelorismo3. Micrognatia4. Clinodactilia del quinto dedo5. Sindactilia | |
| Sección B | Uno de los 3 criterios arriba mencionados, más lo siguiente: Que exista en la familia por lo menos un integrante diagnosticado con 2 de los 3 criterios de la sección A |
| El 60% de los afectados manifiestan la tríada de características cardinales y el 80% presentan 2 de 3 características | |
Desde el punto de vista del diagnóstico molecular, como hasta el día de hoy no se han descrito mutaciones en otro gen distinto al KCNJ2, los individuos con ATS se clasifican como ATS1 si se encuentra alguna mutación en KCNJ2 y como ATS2 si se estudia este gen y no se encuentra ninguna mutación. Es importante aclarar que desde el punto de vista fenotípico, ATS1 y ATS2 son indistinguibles.
Aunque las extrasístoles ventriculares y la taquicardia ventricular no sostenida son muy frecuentes, es menos común la presencia de síncope arrítmico, paro cardíaco o muerte súbita31,32. Esta discordancia entre una carga arrítmica elevada (extrasístoles y taquicardias ventriculares frecuentes) y la aparente baja incidencia de muerte súbita es un asunto que no ha sido explorado en profundidad. En la tabla 2 se mencionan las características ecocardiográficas y electrocardiográficas que se han señalado en el ATS. Aunque la mayoría de los casos se presentan en corazón estructuralmente sano, en algunos se ha observado la presencia de miocardiopatía dilatada33. No se ha definido si esta es una manifestación poco común de la enfermedad o una consecuencia de la taquicardia crónica («taquicardiomiopatía»). Se ha documentado y publicado un caso en el cual la eliminación de la taquicardia ventricular, mediante el uso de flecainida, ha logrado revertir la dilatación ventricular34. Otras características descritas, pero menos comunes en ATS, son las malformaciones cardiovasculares, especialmente de los aparatos valvulares20.
Alteraciones eléctricas y ecocardiográficas descritas en el síndrome de Andersen-Tawil
| Datos electrocardiográficos | Datos ecocardiográficos |
| Onda U prominente o bifásicaIntervalo QTc/Q-Uc prolongadoBigeminismo ventricularExtrasístoles ventricularesTaquicardia ventricularBidireccionalPolimórficaHelicoidalBloqueo auriculoventricularBloqueo de rama (derecha o izquierda)Bloqueo bifascicular | Malformaciones valvularesEstenosis de la pulmonarVálvula aórtica bicúspide, con y sin coartación |
Un dato común en los pacientes con ATS es la presencia de un intervalo QT-U largo (fig. 2), con un patrón característico T-U el cual incluye onda U prolongada, intervalo T-U amplio y onda T terminal prolongada con un declive característico para ATS1 en comparación con los otros SQTL28.
 que salen de la rama ascendente de la onda U. Nótese la prolongación del intervalo QT-U (520ms) a expensas de una onda U (flechas) de gran amplitud y magnitud (véanse las derivaciones V1 a V4). QU corregido (RR 720ms)=612.")
Electrocardiograma de 12 derivaciones en donde se observa ritmo sinusal interrumpido por extrasístoles ventriculares monomórficas (asteriscos) que salen de la rama ascendente de la onda U. Nótese la prolongación del intervalo QT-U (520ms) a expensas de una onda U (flechas) de gran amplitud y magnitud (véanse las derivaciones V1 a V4). QU corregido (RR 720ms)=612.
Como ya se ha señalado, el ATS se hereda de forma autosómica dominante. Al menos el 50% de los individuos diagnosticados con ATS tienen un padre afectado, más del 50% son causados por una mutación de novo. Cada hijo de un individuo con ATS tiene un 50% de probabilidad de heredar la enfermedad (fig. 3); un factor que puede disminuir esta probabilidad es la falta de penetrancia, la cual es evidente en el 6-20% de los individuos con una mutación identificada. Para llevar a cabo el diagnóstico prenatal en una gestante en quien ya se haya identificado la mutación, en ella o en su familia, se puede realizar el análisis genético del producto mediante amniocentesis o biopsia de vellosidades coriales35,36. Hasta la fecha no hay informes de diagnóstico pre-implantacional que podría ser una opción en familias en las que se conoce la mutación.

Árbol genealógico actualizado de la familia mexicana con ATS descrita originalmente por Canún et al.8. Por el tiempo transcurrido, se ha agregado una cuarta generación cuyos miembros tienen el patrón de herencia autosómico dominante.
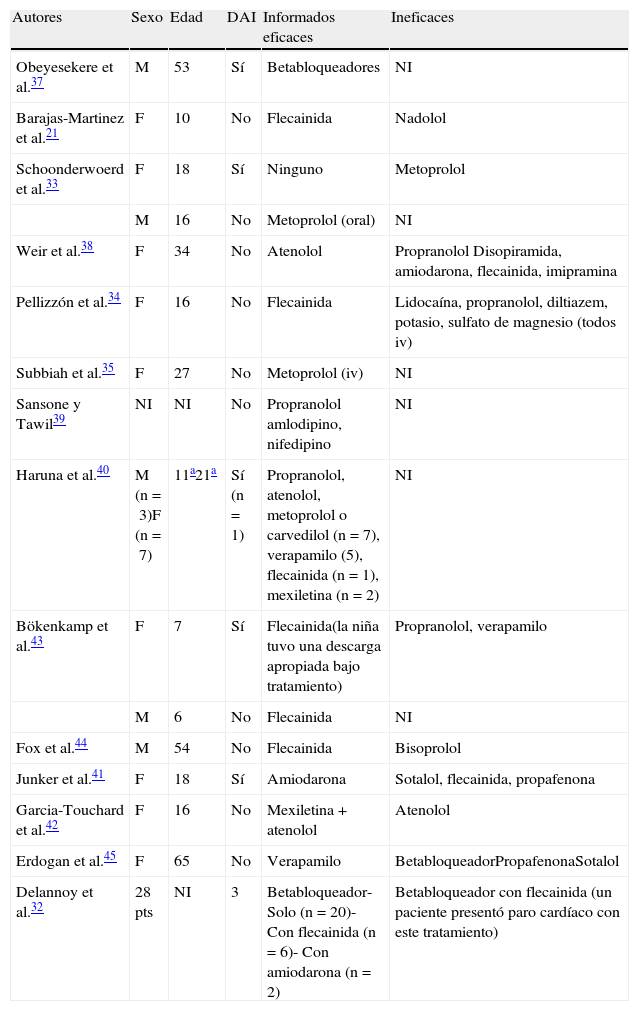
El tratamiento antiarrítmico es controvertido debido a la baja prevalencia de la enfermedad y a la variabilidad en la expresión del fenotipo. Existen pocas publicaciones al respecto y la mayoría son informes de casos aislados o pequeñas series; el objetivo en ellas ha variado, desde disminuir la ectopia ventricular, eliminar la taquicardia ventricular, hasta prevenir la muerte súbita.
En la tabla 3 se presenta un resumen de la literatura. Para la mayoría de los fármacos, los resultados son contradictorios, con informes aislados de su utilidad e informes de su ineficacia37–42. El medicamento que parece ser más útil es la flecainida debido a que hay más publicaciones a favor que en contra de su uso. En casos seleccionados, la flecainida fue muy eficaz para eliminar extrasístoles ventriculares, TV bidireccional e incluso taquicardiomiopatía34,43,44. También se han empleado los betabloqueadores, ya sea solos en combinación con flecainida32 y los calcioantagonistas45 (tabla 3). De cualquier manera, es importante señalar que en la serie de Delannoy et al.32 hubo un caso de paro cardíaco no fatal (gracias a que tenía un cardiodesfibrilador) a pesar del tratamiento con betabloqueador y flecainida.
Fármacos empleados para el tratamiento antiarrítmico en el síndrome de Andersen-Tawil. Se distinguen entre aquellos informados como eficaces o ineficaces
| Autores | Sexo | Edad | DAI | Informados eficaces | Ineficaces |
| Obeyesekere et al.37 | M | 53 | Sí | Betabloqueadores | NI |
| Barajas-Martinez et al.21 | F | 10 | No | Flecainida | Nadolol |
| Schoonderwoerd et al.33 | F | 18 | Sí | Ninguno | Metoprolol |
| M | 16 | No | Metoprolol (oral) | NI | |
| Weir et al.38 | F | 34 | No | Atenolol | Propranolol Disopiramida, amiodarona, flecainida, imipramina |
| Pellizzón et al.34 | F | 16 | No | Flecainida | Lidocaína, propranolol, diltiazem, potasio, sulfato de magnesio (todos iv) |
| Subbiah et al.35 | F | 27 | No | Metoprolol (iv) | NI |
| Sansone y Tawil39 | NI | NI | No | Propranolol amlodipino, nifedipino | NI |
| Haruna et al.40 | M (n=3)F (n=7) | 11a21a | Sí (n=1) | Propranolol, atenolol, metoprolol o carvedilol (n=7), verapamilo (5), flecainida (n=1), mexiletina (n=2) | NI |
| Bökenkamp et al.43 | F | 7 | Sí | Flecainida(la niña tuvo una descarga apropiada bajo tratamiento) | Propranolol, verapamilo |
| M | 6 | No | Flecainida | NI | |
| Fox et al.44 | M | 54 | No | Flecainida | Bisoprolol |
| Junker et al.41 | F | 18 | Sí | Amiodarona | Sotalol, flecainida, propafenona |
| Garcia-Touchard et al.42 | F | 16 | No | Mexiletina+atenolol | Atenolol |
| Erdogan et al.45 | F | 65 | No | Verapamilo | BetabloqueadorPropafenonaSotalol |
| Delannoy et al.32 | 28 pts | NI | 3 | Betabloqueador- Solo (n=20)- Con flecainida (n=6)- Con amiodarona (n=2) | Betabloqueador con flecainida (un paciente presentó paro cardíaco con este tratamiento) |
DAI: desfibrilador automático implantable; Iv: intravenoso; NI: no informado.
Se ha informado de la utilidad de la ablación por radiofrecuencia para el tratamiento de arritmias ventriculares malignas en otros síndromes genéticos como el de Brugada46 por lo que este tratamiento podría ser atractivo en el ATS debido a la frecuente ectopia ventricular. Sin embargo, hasta la fecha no se ha realizado ningún informe sobre ablación exitosa en el ATS3. En una serie de 33 casos con ATS, se informó que se intentó la ablación en 5 casos y en ninguno tuvo éxito32. En una paciente de la serie de ATS del Instituto Nacional de Cardiología se intentó la ablación con catéter en forma infructuosa en 2 ocasiones (datos no publicados).
Al igual que en otros síndromes de muerte súbita, el cardiodesfibrilador automático implantable es una indicación de clase i en aquellos pacientes con paro cardíaco por fibrilación ventricular, síncopes recurrentes o taquicardia ventricular sintomática47.
ConclusiónEl ATS tiene un fenotipo característico por lo que es indispensable que los médicos, en particular los cardiólogos, lo puedan identificar así como que conozcan su importancia desde el punto de vista de su asociación con muerte súbita para poder asesorar a los pacientes y a sus familiares.
FinanciaciónNinguno.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la compañía Sistemas Genómicos® S.L. (Valencia, España) por la secuenciación masiva (next generation sequencing) para la determinación de la mutación KCNJ2 (R218W) de la segunda familia mexicana con ATS reflejada en la figura 1.