








Congestive hepatopathy (CH) is a hepatic vascular disease that results in chronic liver congestion, which can lead to liver fibrosis. New uses of metformin have been discovered over the years. However, the function of metformin in congestive liver fibrosis is not yet fully understood. This study aimed to investigate the effect of metformin on liver fibrosis in a mouse model of CH.
Materials and MethodsPartial ligation of the inferior vena cava (pIVCL) was used to establish a mouse model of liver congestion. Metformin (0.1%) was added to the daily drinking water of the animals, and the effect of metformin on liver tissue was studied after 6 weeks. Hepatic stellate cells (HSCs) were also stimulated with CoCl2 to investigate the inhibitory impact of metformin on the mammalian target of rapamycin (mTOR)/hypoxia-inducible factor-1α (HIF-1α) pathway.
ResultsMetformin attenuated liver congestion; decreased the expression of collagen, fibronectin, α-smooth muscle actin (α-SMA), and HIF-1α; and ameliorated liver fibrosis in pIVCL mice. The proliferation and migration of HSCs were inhibited by metformin in vitro, which prevented α-SMA expression and restrained HSC activation. The expression levels of phosphorylated-mTOR, HIF-1α, and vascular endothelial growth factor were also decreased.
ConclusionsMetformin inhibits CH-induced liver fibrosis. Functionally, this beneficial effect may be the result of inhibition of HSC activation and of the mTOR/HIF-1α signaling pathway.
Congestive hepatopathy (CH) is an obstructive lesion above the opening of the hepatic vein that can be caused by various reasons, including the Budd-Chiari syndrome and right-sided heart failure [1]. Outflow tract obstruction can lead to hepatic venous congestion, subsequently leading to ischemia-hypoxic injury of adjacent hepatocytes [2]. Chronic hepatic congestion leads to increased sinusoidal pressure and dilation, sinusoidal thrombosis, and hepatic fibrosis [3]. The progression of liver fibrosis is accompanied by an accumulation of extracellular matrix (ECM) and collagen. Liver fibrosis can convert to cirrhosis where the rate of ECM degradation is less than that of its synthesis, and even progress to hepatocellular carcinoma [4].
Hypoxia is a significant process in the pathogenesis of liver disease [5], and it may be one of the determinants of liver disease in various pathological conditions. Cells respond to hypoxia through activation of hypoxia-inducible factor (HIF) transcription factors. A variety of oxygen-regulated transcription factors control the gene expression changes induced by hypoxia, among which the mammalian target of rapamycin (mTOR) and HIF-1 signaling pathways are of significance. Hypoxia upregulates mTOR and activates its downstream target genes, whereas HIF-1α affects hepatic stellate cell (HSC) activation and collagen deposition, induces angiogenesis and epithelial-mesenchymal transition, and mediates gene modification in liver cirrhosis [6].
Metformin, as a classic drug in the treatment of diabetes, has been widely used in the clinic over the years [7]. Besides its hypoglycemic effects, increasing research suggests that metformin has various other functions, including functioning as an antioxidant [8–10]. A previous study [11] indicated that metformin can inhibit the TGF-β1/Smad3 pathway and improve liver fibrosis induced by carbon tetrachloride (CCl4). In nonalcoholic steatohepatitis (NASH) [12], the use of metformin has been associated with improved liver condition. Congestive hepatic fibrosis mainly develops in the central part of the hepatic lobules, although this pattern of fibrosis is not unique to CH and can also be observed in chronic alcoholic injury, NASH, and CCl4-induced liver injury [13]. However, RNA expression of fibrogenesis and angiogenic factors in CH is distinct from that in chronic liver disease associated with alcoholic or viral hepatitis [14]. In our study, we developed a CH mouse model by partial ligation of the inferior vena cava (pIVCL), to explore the relationship between metformin and congestive hepatic fibrosis as well as the potential underlying mechanisms.
2Materials and Methods2.1MaterialsMetformin and cobalt chloride hexahydrate (CoCl2·6H2O) were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2-Methoxyestradiol (2-MeOE2) and rapamycin were purchased from MCE (Monmouth Junction, NJ, USA). The rabbit anti-mouse phosphorylated-mTOR (p-mTOR), mTOR, and HIF-1α antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). GAPDH, collagen I (Col I), fibronectin (FN), α-smooth muscle actin (α-SMA), and vascular endothelial growth factor (VEGF) antibodies were obtained from Abcam (Cambridge, UK). The goat anti-rabbit antibody and the enhanced chemiluminescence (ECL) reagents were supplied by Cwbio (Jiangsu, China).
2.2AnimalsFour-week-old male C57BL/6J mice (20–22 g) were obtained from the Experimental Animal Center of Zhengzhou University. The mice were fed in individually ventilated cages at 22℃ in 12-h light-dark cycles, with free access to adequate food and water. All mice were acclimatized for at least 1 week before experiments. Animals were randomly divided into four groups: sham-operated group (SHAM, n = 10), sham-operated + metformin group (MET, n = 10), pIVCL group (pIVCL, n = 10), and pIVCL + metformin group (pIVCL+MET, n = 10). pIVCL was performed for disease modeling, [3] as described. Under sterile conditions and isoflurane inhalation anesthesia, an incision was made below the xiphoid process of the mice; the liver and diaphragm were separated, and the suprahepatic inferior vena cave (IVC) was exposed. The IVC and wire with a diameter of 0.6 mm were initially ligated together with a silk thread, and then the wire was gently removed. The SHAM and MET groups underwent all the steps of pIVCL except for the ligation. The drinking water of the MET and pIVCL+MET groups was infused with 0.1% metformin daily. Mice were sacrificed by cervical dislocation after 6 weeks, and the liver samples were acquired for subsequent evaluation. Serum samples were used to evaluate liver function using an assay kit (Solarbio, Beijing, China).
2.3Pathological staining analysis and immunohistochemistry (IHC)After 6 weeks, we calculated the proportion of liver weight to body weight, which represents the liver index. Liver tissue was fixed in 4% paraformaldehyde, embedded in paraffin, and sliced into 4 μm thick sections that were mounted on slides. The slides were stained with hematoxylin and eosin (H&E) and sirius red (SR), according to standard procedures. FN and α-SMA were detected by IHC according to the manufacturer's instructions (Cwbio, Jiangsu, China). SR staining, FN and α-SMA positive samples were evaluated using the ImageJ software.
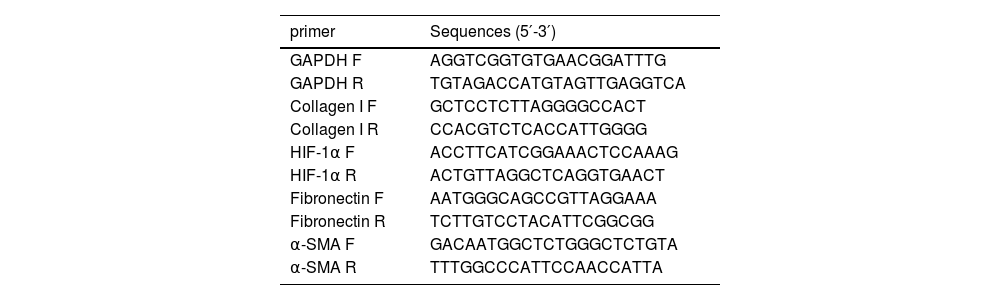
2.4Quantitative Real-Time Polymerase Chain Reaction (qPCR)The Trizol reagent (Cwbio, Jiangsu, China) was used to extract total RNA from liver tissue, and a reverse transcription kit (Vazyme, Nanjing, China) was used to reverse transcribe the total RNA into cDNA. Amplifications were detected on a Real-Time PCR system (Thermo Fisher, USA) using SYBR qPCR MasterMix (Vazyme, Nanjing, China). The expression of GAPDH in the same sample was used as the internal reference, and the relative expression of genes was calculated using the 2−ΔΔCt formula. The primers used in this study are listed in Table 1.
Primer sequences.
F, forward primer; R, reverse primer.
The human HSC line LX-2 was obtained from icellbio (Shanghai, China). LX-2 cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum.
2.6Proliferation assayA total of 5×103 LX2 cells were seeded in 96well plates and incubated for 24 h. Following this, the culture medium was changed to medium containing CoCl2·6H2O (150 μM) or different concentrations of metformin, and incubation was continued for 24 and 48 h. At the endpoint of the assay 10 μl of CCK8 was added to each well. After 2 h, the optical density values were measured with a spectrophotometer at 450 nm for cell proliferation analysis.
2.7Cell migration assessmentA total of 5×105 LX-2 cells were seeded in 6-well plates and incubated overnight. Next, the culture medium was switched to medium without serum for 8 h. A scratch was made in each well using a pipette tip, and plates were washed with phosphate-buffered saline. CoCl2·6H2O and metformin (1 mM, 5 mM) were added to the respective wells. Images were captured at 0 and 24 h.
2.8Western blot analysisMouse liver tissues and LX-2 cell line protein lysates were prepared using RIPA lysis buffer containing protease and phosphatase inhibitors, on ice. Protein concentrations were measured using the bicinchoninic acid method. After adding the protein samples to the corresponding sample wells, electrophoresis was performed with SDS-PAGE, and the resolved proteins were transferred to PVDF membranes. The membranes were then blocked with 5% skimmed milk for 1 h and incubated with the corresponding primary antibodies overnight at 4℃. Subsequently, the membranes were washed three times with TBST, incubated with a secondary antibody for 1 h at 37℃, and washed again with TBST. Protein bands were detected by using ECL reagents.
2.9Statistical analysisData are presented as means ± standard deviations. Statistical significance was determined using oneway analysis of variance followed by Turkey's post hoc test. A value of p < 0.05 was considered statistically significant.
2.10Ethical statementAll animal experiments complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and were conducted in accordance with the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines, EU Directive 2010/63/EU for Animal Experiments. This study was approved by the Ethics Committee of Zhengzhou University (KY-2021-00911).
3Results3.1Metformin inhibited liver fibrosis and ameliorated liver function in pIVCL miceAfter 6 weeks of pIVCL treatment, the mice developed liver fibrosis. The degree of fibrosis was not significant in the SHAM and MET mice. In the pIVCL mice, ascites and abdominal wall varicose veins were observed, and congestion and sclerosing nodules appeared in the liver, which were alleviated by metformin treatment. In the pIVCL group, H&E staining showed congestion in the hepatic sinusoids adjacent to the central hepatic vein, disordered hepatocyte arrangement, hepatic sinusoids dilatation, and a few small clusters of inflammatory cells. The use of metformin reduced congestion (Fig. 1 A). Collagen was measured using SR staining in the liver tissue, which showed collagen to be mainly distributed in zone 3 rather than in the portal zone, and interconnected to form bridging fibrosis. Metformin inhibited collagen deposition (Fig. 1 A, B). Liver index analysis showed that the increased liver index observed in the pIVCL group was reduced by metformin (Fig. 1 C). Finally, we used a liver function assay kit to measure the serological levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The results indicated that metformin could improve the abnormal liver function caused by pIVCL (Fig. 1 D).
 H&E and SR staining of liver tissues after 6 weeks of pIVCL; black arrow: hepatic sinusoid dilatation; white arrow: disordered hepatocyte arrangement; black circle: congestion in the hepatic sinusoid; white circle: a small cluster of inflammatory cells (H&E: 200× magnification, scale = 50μm; SR: 100× magnification, scale = 100μm); (B) proportion of positive area of SR staining; (C) data of liver index (the ratio of liver weight to body weight); (D) MET decreased the serum ALT and AST levels. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. n = 10/group. MET, metformin; pIVCL, partial ligation of the inferior vena cava; H&E, hematoxylin and eosin; SR, sirius red; ALT, alanine aminotransferase; AST, aspartate aminotransferase.")
MET attenuated liver congestion and reduced deposition of collagen. (A) H&E and SR staining of liver tissues after 6 weeks of pIVCL; black arrow: hepatic sinusoid dilatation; white arrow: disordered hepatocyte arrangement; black circle: congestion in the hepatic sinusoid; white circle: a small cluster of inflammatory cells (H&E: 200× magnification, scale = 50μm; SR: 100× magnification, scale = 100μm); (B) proportion of positive area of SR staining; (C) data of liver index (the ratio of liver weight to body weight); (D) MET decreased the serum ALT and AST levels. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. n = 10/group. MET, metformin; pIVCL, partial ligation of the inferior vena cava; H&E, hematoxylin and eosin; SR, sirius red; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Next, we assessed the expression of FN, which is one of the essential components of ECM. IHC revealed that FN was highly expressed in the pIVCL group (Fig. 2 A, B). At the mRNA level, the expression of FN and Col I increased after mice were subjected to pIVCL; however, metformin treatment decreased FN and Col I mRNA expression (Fig. 2 C, D). The western blot results showed that metformin reduced the protein levels of FN and Col I (Fig. 2 E), which was consistent with the qPCR data. Thus, metformin alleviated the pIVCL-induced increase in FN levels and collagen deposition.
 FN expression levels in the liver tissue detected by immunohistochemistry (100× magnification, scale = 100μm); (B) proportion of positive area for FN; (C-D) FN and Col I mRNA expression levels detected by PCR; (E) FN and Col I protein levels detected by western blot. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. n = 10/group. MET, metformin; pIVCL, partial ligation of the inferior vena cava; Col I, collagen I; FN, fibronectin.")
Effects of MET on FN and Col I. (A) FN expression levels in the liver tissue detected by immunohistochemistry (100× magnification, scale = 100μm); (B) proportion of positive area for FN; (C-D) FN and Col I mRNA expression levels detected by PCR; (E) FN and Col I protein levels detected by western blot. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. n = 10/group. MET, metformin; pIVCL, partial ligation of the inferior vena cava; Col I, collagen I; FN, fibronectin.
We observed that α-SMA was highly expressed in the pIVCL group, it mainly clustered near the central hepatic vein, and arranged radially around the hepatic sinusoids (Fig. 3 A, B). Metformin inhibited the deposition of α-SMA. At the tissue level, the PCR and western blot analysis confirmed the overexpression of HIF-1α and α-SMA in hepatic congestion; meanwhile, metformin exerted an antagonistic effect on HIF-1α and α-SMA expression (Fig. 3 C, D, E).
 expression levels of α-SMA in the liver tissues detected by immunohistochemistry (200× magnification, scale = 50μm); (B) proportion of positive area of α-SMA; (C-D) mRNA expression levels of HIF-1α and α-SMA detected by qPCR; (E) protein expressions of HIF-1α and α-SMA detected by western blot. n = 10/group. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. MET, metformin; pIVCL, partial ligation of the inferior vena cava; α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor-1α.")
MET decreased expression of α-SMA and HIF-1α: (A) expression levels of α-SMA in the liver tissues detected by immunohistochemistry (200× magnification, scale = 50μm); (B) proportion of positive area of α-SMA; (C-D) mRNA expression levels of HIF-1α and α-SMA detected by qPCR; (E) protein expressions of HIF-1α and α-SMA detected by western blot. n = 10/group. #p < 0.05, vs. pIVCL; * p < 0.01, vs. pIVCL. MET, metformin; pIVCL, partial ligation of the inferior vena cava; α-SMA, α-smooth muscle actin; HIF-1α, hypoxia-inducible factor-1α.
We continued by investigating the properties of HSC under hypoxia. We used CoCl2 to simulate an anoxic environment and treated HSC with different concentrations of metformin for 24 h (Fig. 4 A). Metformin restrained the proliferation of HSC in a dose-dependent manner, with an IC50 of 51.69 mmol/L. We selected two safe concentrations (1 mM and 5 mM) to explore the effect of metformin on HSC under CoCl2 stimulation. As shown in Fig. 4 B, CoCl2 inhibited HSC proliferation, which was not ameliorated by metformin. This inhibition was exacerbated at relatively high metformin doses (5 mM). The effect of metformin on the migration of HSC was investigated by the scratch test. The results (Figure 4 C, D) indicated that CoCl2 stimulated the migration of HSCs, and concomitant treatment with metformin prevented this effect; the migration-inhibitory effect of metformin was more pronounced at higher concentrations (5 mM).
 CCK8 assay used to measure the effect of MET on the proliferation of HSCs. HSCs were cultured in different concentrations of MET (1, 5, 10, 20, 50, 100 mM) for 24 h; (B) HSCs cultured with or without MET (1, 5 mM) and CoCl2 (150 μM). Cell proliferation was measured at 24 and 48h; (C) Scratch test was used to evaluate cell migration. Images (100× magnification, scale = 100 μm) were obtained at 0 and 24 h; (D): ImageJ was used to measure the distance from the scratch edge to calculate the migration ability. n = 3. #p < 0.05, vs. CoCl2 only treatment; * p < 0.01, vs. CoCl2 only treatment. MET, metformin; HSC, hepatic stellate cell; CoCl2, cobalt chloride.")
Effect of MET on proliferation and migration of HSCs: (A) CCK8 assay used to measure the effect of MET on the proliferation of HSCs. HSCs were cultured in different concentrations of MET (1, 5, 10, 20, 50, 100 mM) for 24 h; (B) HSCs cultured with or without MET (1, 5 mM) and CoCl2 (150 μM). Cell proliferation was measured at 24 and 48h; (C) Scratch test was used to evaluate cell migration. Images (100× magnification, scale = 100 μm) were obtained at 0 and 24 h; (D): ImageJ was used to measure the distance from the scratch edge to calculate the migration ability. n = 3. #p < 0.05, vs. CoCl2 only treatment; * p < 0.01, vs. CoCl2 only treatment. MET, metformin; HSC, hepatic stellate cell; CoCl2, cobalt chloride.
α-SMA is a marker of HSC activation. Metformin decreased the levels of α-SMA in CoCl2-induced HSCs (Fig. 5 A), indicating its role in suppressing HSC activation. We then explored the expression levels of components of the mTOR/HIF-1α signaling pathway using western blot. Metformin reduced the CoCl2-induced expression of p-mTOR, HIF-1α, and VEGF (Fig. 5 B). These results suggested that metformin is related to the mTOR/HIF-1α signaling pathway. Furthermore, we treated activated HSCs with 2-MeOE2 (HIF-1α inhibitor) and found that it could reduce the expression of p-mTOR, HIF-1α, and VEGF. Rapamycin (mTOR inhibitor) also showed similar results (Fig. 5 B). Collectively, these observations suggest the involvement of the mTOR/HIF-1α signaling pathway in the activation of HSCs.
, MET (5 mM), 2-MeOE2 (10 μM), and rapamycin (100 nM): (A) Western blot analysis was used to detect the expression of α-SMA; (B) p-mTOR, mTOR, HIF-1α, and VEGF expressions were measured by Western blot analysis. n = 3. #p < 0.05, vs. CoCl2 only treatment; * p < 0.01, vs. CoCl2 only treatment. MET, metformin; HSC, hepatic stellate cell; CoCl2, cobalt chloride; 2-MeOE2, 2-Methoxyestradiol; α-SMA, α-smooth muscle actin; p-mTOR, phosphorylated-mammalian target of rapamycin; mTOR, mammalian target of rapamycin; HIF-1α, hypoxia-inducible factor-1α; VEGF, vascular endothelial growth factor.")
Effect of MET on α-SMA and mTOR/HIF-1α pathway in HSC. Cells were cultured with or without CoCl2 (150 μM), MET (5 mM), 2-MeOE2 (10 μM), and rapamycin (100 nM): (A) Western blot analysis was used to detect the expression of α-SMA; (B) p-mTOR, mTOR, HIF-1α, and VEGF expressions were measured by Western blot analysis. n = 3. #p < 0.05, vs. CoCl2 only treatment; * p < 0.01, vs. CoCl2 only treatment. MET, metformin; HSC, hepatic stellate cell; CoCl2, cobalt chloride; 2-MeOE2, 2-Methoxyestradiol; α-SMA, α-smooth muscle actin; p-mTOR, phosphorylated-mammalian target of rapamycin; mTOR, mammalian target of rapamycin; HIF-1α, hypoxia-inducible factor-1α; VEGF, vascular endothelial growth factor.
Liver fibrosis is a crucial phase in the development of liver disease mostly characterized by excessive deposition of ECM, and early intervention is of great importance for preventing the development of liver cirrhosis and liver cancer [15]. A previous study [11] reported that metformin can inhibit the TGF-β1/Smad3 pathway to improve CCl4-induced liver fibrosis. Furthermore, Li et al. [16] reported that metformin can reduce the expression of HIF-1α in the CCl4 model and play a role by modulating the AKT/mTOR pathway. In NASH-induced liver fibrosis [12], metformin, as an activator of the adenosine monophosphate-activated protein kinase (AMPK), can improve HSC activation by inhibiting the succinate-GPR91 pathway. She et al. [17] found that knockdown of HIF-1α can reduce the expression of TGF-β1 and α-SMA in HSCs. This indicates that different types of liver fibrosis have similar mechanisms. In this study we found high expression of HIF-1α and α-SMA in the liver of pIVCL mice, indicating that hypoxia plays a role in congestive hepatic fibrosis. In fact, hypoxia is a common process in many chronic liver diseases. Here, we observed that the liver of pIVCL mice was clearly congested, and extensive erythrocyte sedimentation near the central vein could be seen by H&E staining. This phenomenon, however, was not observed in liver fibrosis induced by CCl4 and NASH. Obstruction in the hepatic venous outflow tract causes hepatic venous pressure to be transmitted to the central hepatic vein resulting in increased sinusoidal pressure, which decreases the production of vasodilator by sinusoidal endothelial cells and leads to vasoconstriction. Vascular constriction and congestion lead to hypoxic injury of adjacent hepatocytes [18] and to hepatic sinusoidal dilatation. Over time, liver fibrosis develops gradually around the central vein (zone III) and forms bridging liver fibrosis [19]. Under metformin treatment, liver congestion was alleviated, and liver fibrosis was improved, indicating that one of the roles of metformin is related to the reduction of liver congestion.
HSC activation is generally considered to be the central link in liver fibrosis [20]. When simulating various pathogenic factors, inactive HSCs differentiate into proliferative, migratory, and contractile myofibroblasts and secrete ECM [21]. α-SMA is the indicator of HSC activation, and its level denotes the quantification and identification of activated HSCs in the progression of liver cirrhosis [22]. Activated HSCs have the ability to contract, leading to constriction of hepatic sinusoids, restraining blood flow to the liver, and exacerbating hypoxia [6]. We found that α-SMA was highly expressed in the pIVCL group, and IHC showed that it was deposited adjacent to the central hepatic vein. Metformin decreased α-SMA expression, suggesting that its effect may be related to HSCs.
Under normal circumstances, in a static state, HSCs exist in the Disse gap and sinuses. The chemotaxis of activated HSCs, however, can be enhanced by chemokines, and can manifest as increased motility [23]. We further explored possible signaling mechanisms involved in this process. The mTOR and AMPK signaling pathways are involved in the progression of liver fibrosis and are crucial factors in regulating intracellular signal transduction [24]. Many studies have shown that there is an interaction between the PI3K/mTOR and MAPK/ERK pathways, which together regulate HSC differentiation and proliferation [16,25]. As a downstream molecule of the mTOR pathway, HIF-1α can upregulate the levels of VEGF, platelet-derived growth factor (PDGF), and tissue inhibitor of metalloproteinase (TIMPs), which play significant roles in the activation of HSCs [26–28]. We used CoCl2 to simulate a hypoxic environment for stimulating HSCs. In that setting, the phosphorylation levels of mTOR increased, which led to an increase in the levels of HIF-1α and VEGF under hypoxia. By contrast, metformin decreased the expression levels of p-mTOR, HIF-1α, and VEGF. Previous studies [16] have shown that metformin reduced the expression of p-mTOR by activating AMPK, which is consistent with our results. This suggested that metformin may inhibit the mTOR/HIF-1α pathway through AMPK, thereby inhibiting HSC activation. We treated HSCs with rapamycin (mTOR inhibitor) and found that rapamycin could inhibit the expression of p-mTOR, HIF-1α, and VEGF. Interestingly, after treating HSCs with 2-MeOE2 (HIF-1α inhibitor) we observed that in addition to the decreased expression of HIF-1α and its downstream target VEGF, expression of the upstream p-mTOR was also decreased. Given that 2-MeOE2 cannot directly act on mTOR, we speculated that its effect may be related to the improvement of hypoxia in HSCs and the decrease of HSC activation, resulting in a decrease in mTOR pathway activation.
5ConclusionsIn conclusion, therapy with metformin while establishing the pIVCL mouse model can alleviate hepatic fibrosis through inhibition of HSC activation and inhibition of the mTOR/HIF-1α signaling pathway. This provides a potential possibility for the clinical treatment of congestive liver fibrosis with metformin. Whether or not metformin can reverse liver fibrosis and the precise mechanisms underlying this process remain unclear, and need to be further investigated.
Jing Yang: Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Writing – original draft. Suxin Li: Funding acquisition, Project administration, Resources, Validation, Writing – review & editing. Shengyan Liu: Software, Validation, Data curation. Yuehui Zhang: Formal analysis, Methodology, Visualization. Dongqi Shen: Conceptualization, Investigation, Supervision, Validation. Peiju Wang: Formal analysis, Project administration, Validation. Xiaowei Dang: Conceptualization, Data curation, Funding acquisition, Project administration, Supervision, Writing – review & editing.



