

Hasta hace 30 años, cuando la cirugía era el único tratamiento disponible, el 90% de los pacientes afectados de OS moría en el primer año tras el diagnóstico, a pesar del control local de la enfermedad, lo que indicaba que en el momento del diagnóstico la mayoría de los pacientes habían desarrollado micrometástasis a distancia1. con los protocolos actuales, que combinan la QT y la cirugía, la supervivencia libre de enfermedad (SLE) a los 10 años se sitúa en torno al 60% en los casos más favorables2.
EpidemiologíaEl OS es el tumor óseo primario más frecuente en la infancia y la adolescencia (56% de los tumores óseos, 5% de los cánceres pediátricos). Su incidencia es de 3 nuevos casos por millón de personas por año. El pico medio de edad de presentación es a los 16 años; predomina en varones frente a mujeres en una proporción de 1,6:13.
Histopatogenia y etiologíaEl OS se caracteriza por la producción de sustancia osteoide y hueso por una célula mesenquimal maligna de la estroma. La Organización Mundial de la Salud reconoce 3 tipos convencionales: osteoblástico (50%), fibroblástico (25%) y condroblástico (25%), en función del tipo predominante de matriz que produzcan. Se reconocen 4 variantes: la telangiectásica (quístico, vascularizado), multifocal, histiocitoma fibroso maligno y el de célula pequeña, altamente agresivo, y morfológicamente similar al SEw4.
Entre los posibles agentes etiológicos, la exposición a radiación ionizante es el único factor exógeno probado, con períodos de inducción entre 10 y 20 años5. La administración previa de QT, particularmente agentes alquilantes, se ha relacionado también, y podría potenciar el efecto de la RT6. Sin embargo, en la mayoría de los pacientes no se reconocen estos antecedentes ni predisposición familiar. Se han descrito asociaciones con enfermedades hereditarias poco frecuentes, como el retinoblastoma hereditario bilateral, los síndromes de Li-Fraumeni, Rothmund-Thomson o de Bloom, o con la exostosis múltiple y la enfermedad de Paget6,7.
Hay una serie de alteraciones genéticas asociadas con el OS; la más destacada es la del gen RB, en el cromosoma 13 (gen supresor tumoral del retinoblastoma, codificador de una proteína nuclear que inhibe el crecimiento celular). Los pacientes afectados de retinoblastoma presentan un incremento en la incidencia de segundos tumores, especialmente de OS (hasta 500 veces más). Se ha detectado una incidencia elevada de mutaciones homocigotas del gen RB en células de OS. Otra alteración presente es la mutación homocigota del gen p53, relacionada con el control del crecimiento y del ciclo celular7–9.
Se ha descartado el antecedente de un traumatismo como agente etiológico del OS; se trata de un factor que pone de manifiesto la enfermedad, pero que no la genera.
El OS es un tumor típico del hueso en crecimiento. Este hecho se apoya en que el OS es más frecuente en los huesos largos, como fémur y tibia, en las metáfisis, en los momentos de mayor desarrollo (el estirón puberal), en el sexo con mayor crecimiento (varones) y en los individuos más altos; aparece de forma más temprana en las mujeres, como consecuencia de su estirón puberal más temprano10.
ClínicaEl dolor suele ser el síntoma más temprano -aparece hasta 4 meses antes del diagnóstico-, desencadenado en ocasiones por ejercicio físico o traumatismos (es posible la aparición de fracturas patológicas). Con el tiempo aparecerá inflamación local y efecto de masa. La sintomatología sistémica no es frecuente en la enfermedad localizada.
En el momento del diagnóstico, el OS se presenta como enfermedad metastásica en el 20%. El 80% de los OS surge en las extremidades (fémur, 40%; tibia, 20%; húmero, 10%), y crece desde la cavidad medular hacia la corteza y los tejidos blandos. El 20% restante aparece en el esqueleto axial. El lugar donde más frecuentemente metastatiza es el pulmón, seguido del hueso; son raras otras localizaciones11.
LaboratorioLos hallazgos más destacables son el incremento de la fosfatasa alcalina (que en ausencia de enfermedad metastásica se correlaciona con la masa tumoral), el incremento de la lactatodeshidrogenasa (LDH) y de la velocidad de sedimentación globular (VSG). Es posible hallar una anemia moderada de trastornos crónicos7,12.
RadiologíaLa aproximación inicial al diagnóstico debe hacerse mediante pruebas de imagen. En la radiografía simple aparece como una masa con regiones osteolíticas y escleróticas, de bordes mal definidos, que suele originarse en la cavidad medular y progresa hacia la corteza, atravesando y levantando el periostio (que reacciona formando tejido óseo inmaduro, en forma de triángulo: signo de Codman), y que puede afectar a los tejidos blandos que rodean al hueso, produciendo imágenes difusas, de diferentes densidades (fig. 1). Sin embargo, es necesario realizar pruebas más sensibles, como la resonancia magnética (RM) o la tomografía computarizada (TC) con contraste para determinar la afectación nerviosa, vascular y de tejidos blandos, así como para precisar la extensión del tumor en el propio hueso. La RM es superior a la TC para delimitar la extensión del tumor, la afectación de los paquetes neurovasculares, la articulación y la médula ósea, así como para detectar la existencia de lesiones satélites13 (fig. 2). El papel de la tomografía por emisión de positrones (PET) no está completamente definido en pacientes con OS14.


Lectura rápida

Los tumores óseos malignos suponen el 6% de todas las neoplasias infantiles, entre los cuales los más frecuentes son el osteosarcoma (OS) y los tumores de la familia Ewing (TFEw). Ambos son muy agresivos y tienen gran tendencia a metastatizar.
OsteosarcomaEl OS es el tumor óseo primario más frecuente en los niños. Se relaciona con la exposición a la radiación ionizante, y se asocia a ciertas enfermedades congénitas poco frecuentes.
ClínicaEl síntoma inicial es el dolor en la región afectada, y con el tiempo aparecerá la inflamación local. El 80% de los OS surgen en las extremidades, y el 20% de los pacientes tiene metástasis al diagnóstico en pulmón y huesos. Radiológicamente, aparece como una masa con regiones osteolíticas y escleróticas, que levanta y rompe el periostio. Con la resonancia magnética (RM) y la tomografía computarizada (TC) se establecen la afectación nerviosa, vascular, de tejidos blandos y la extensión del tumor en el propio hueso.

Lectura rápida
El diagnóstico es histológico, con una biopsia realizada preferentemente con un trócar; el estudio de extensión incluirá una gammagrafía ósea con tecnecio 99 y una TC torácica.
PronósticoLa supervivencia global se sitúa en torno al 70%, y son factores de mal pronóstico la presencia de metástasis al diagnóstico y la localización del tumor en el esqueleto axial.
TratamientoLa cirugía es la pieza clave del tratamiento, pero por sí sola no es capaz de curar la enfermedad, dada la alta incidencia de micrometástasis desde el inicio. Los protocolos de tratamiento incluyen quimioterapia neoadyuvante (preoperatoria), seguida de cirugía, con lo que se intenta resecar completamente el tumor en bloque; el sistema musculoesquelético se reconstruye mediante injertos óseos o prótesis. Tras la cirugía, se reanudan los ciclos de quimioterapia.
El diagnóstico de certeza debe apoyarse en la confirmación histopatológica. Se recomienda que la biopsia la realice un cirujano ortopédico con experiencia en tumores óseos, preferiblemente el mismo que realizará la extirpación del tumor y la reconstrucción osteomuscular. Es preferible que se realice con trócar dentro de la zona que después será resecada, cuidando que no se produzcan fracturas patológicas o contaminación tumoral de los tejidos circundantes.
Estudio de extensión tumoralEs preciso realizar una gammagrafía ósea con fosfonatos marcados con tecnecio 99 (99Tc), ya que ésta es la prueba que mejor evalúa el esqueleto para detectar metástasis óseas a distancia y lesiones satélites. Dada la posibilidad de enfermedad diseminada al diagnóstico, y dado que el lugar de metástasis más frecuente son los pulmones, se debe realizar una TC torácica. Las metástasis pulmonares aparecen en localización subpleural, redondeadas, densas y calcificadas. Dada la posibilidad de resultados falsos-positivos, en casos de duda es necesaria la confirmación histológica15.
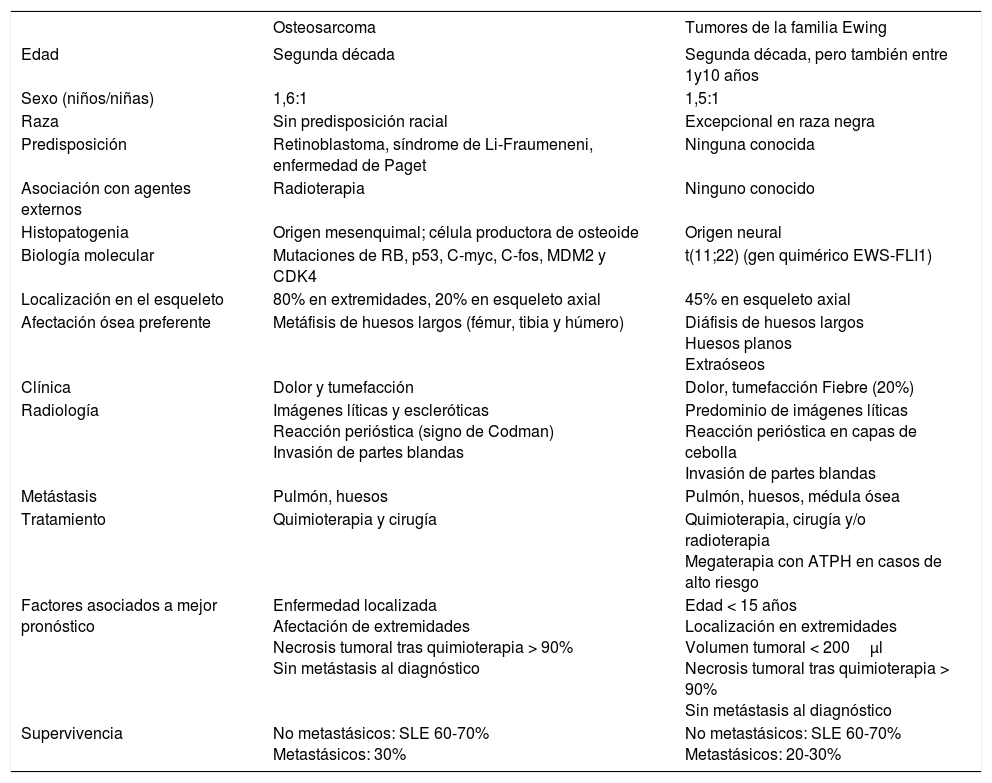
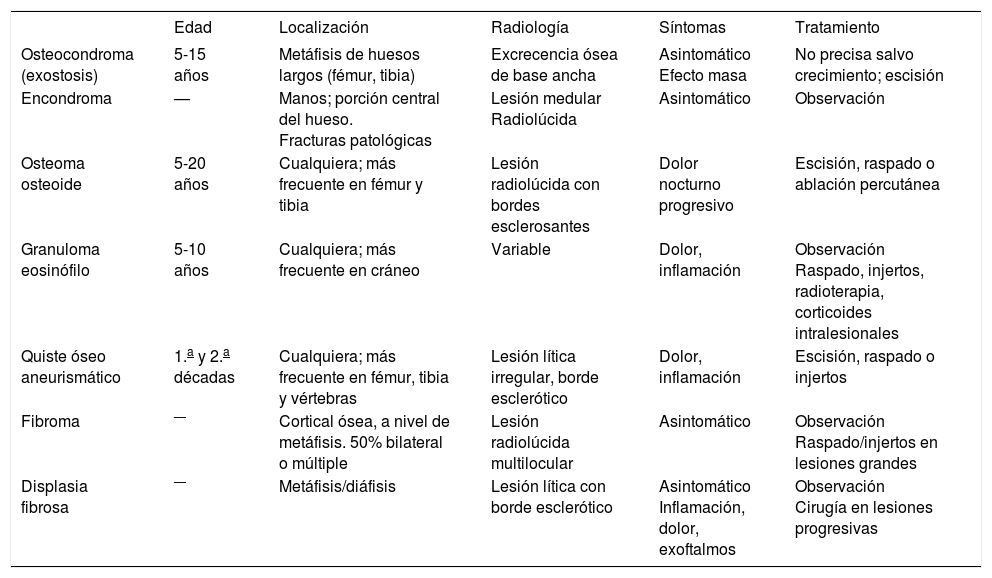
Diagnóstico diferencialPrincipalmente ha de hacerse frente a los tumores de la familia Ewing (TFEw) (tabla 1) y con los tumores óseos benignos (tabla 2). El diagnóstico histopatológico bastará para diferenciar estas entidades.
Comparación de hallazgos entre osteosarcoma y tumores de la familia Ewing
| Osteosarcoma | Tumores de la familia Ewing | |
|---|---|---|
| Edad | Segunda década | Segunda década, pero también entre 1y10 años |
| Sexo (niños/niñas) | 1,6:1 | 1,5:1 |
| Raza | Sin predisposición racial | Excepcional en raza negra |
| Predisposición | Retinoblastoma, síndrome de Li-Fraumeneni, enfermedad de Paget | Ninguna conocida |
| Asociación con agentes externos | Radioterapia | Ninguno conocido |
| Histopatogenia | Origen mesenquimal; célula productora de osteoide | Origen neural |
| Biología molecular | Mutaciones de RB, p53, C-myc, C-fos, MDM2 y CDK4 | t(11;22) (gen quimérico EWS-FLI1) |
| Localización en el esqueleto | 80% en extremidades, 20% en esqueleto axial | 45% en esqueleto axial |
| Afectación ósea preferente | Metáfisis de huesos largos (fémur, tibia y húmero) | Diáfisis de huesos largos Huesos planos Extraóseos |
| Clínica | Dolor y tumefacción | Dolor, tumefacción Fiebre (20%) |
| Radiología | Imágenes líticas y escleróticas Reacción perióstica (signo de Codman) Invasión de partes blandas | Predominio de imágenes líticas Reacción perióstica en capas de cebolla Invasión de partes blandas |
| Metástasis | Pulmón, huesos | Pulmón, huesos, médula ósea |
| Tratamiento | Quimioterapia y cirugía | Quimioterapia, cirugía y/o radioterapia Megaterapia con ATPH en casos de alto riesgo |
| Factores asociados a mejor pronóstico | Enfermedad localizada Afectación de extremidades Necrosis tumoral tras quimioterapia > 90% Sin metástasis al diagnóstico | Edad < 15 años Localización en extremidades Volumen tumoral < 200μl Necrosis tumoral tras quimioterapia > 90% Sin metástasis al diagnóstico |
| Supervivencia | No metastásicos: SLE 60-70% Metastásicos: 30% | No metastásicos: SLE 60-70% Metastásicos: 20-30% |
UATPH: trasplante autólogo de progenitores hematopoyéticos; SLE: superviviencia libre de enfermedad.
Tumores óseos benignos más frecuentes en la infancia y sus características clinicorradiológicas
| Edad | Localización | Radiología | Síntomas | Tratamiento | |
|---|---|---|---|---|---|
| Osteocondroma (exostosis) | 5-15 años | Metáfisis de huesos largos (fémur, tibia) | Excrecencia ósea de base ancha | Asintomático Efecto masa | No precisa salvo crecimiento; escisión |
| Encondroma | — | Manos; porción central del hueso. Fracturas patológicas | Lesión medular Radiolúcida | Asintomático | Observación |
| Osteoma osteoide | 5-20 años | Cualquiera; más frecuente en fémur y tibia | Lesión radiolúcida con bordes esclerosantes | Dolor nocturno progresivo | Escisión, raspado o ablación percutánea |
| Granuloma eosinófilo | 5-10 años | Cualquiera; más frecuente en cráneo | Variable | Dolor, inflamación | Observación Raspado, injertos, radioterapia, corticoides intralesionales |
| Quiste óseo aneurismático | 1.a y 2.a décadas | Cualquiera; más frecuente en fémur, tibia y vértebras | Lesión lítica irregular, borde esclerótico | Dolor, inflamación | Escisión, raspado o injertos |
| Fibroma | — | Cortical ósea, a nivel de metáfisis. 50% bilateral o múltiple | Lesión radiolúcida multilocular | Asintomático | Observación Raspado/injertos en lesiones grandes |
| Displasia fibrosa | — | Metáfisis/diáfisis | Lesión lítica con borde esclerótico | Asintomático Inflamación, dolor, exoftalmos | Observación Cirugía en lesiones progresivas |
Los factores pronósticos significativos han sido los siguientes16:
- —
Enfermedad localizada frente a enfermedad metastásica (SLE a los 5 años del 60-70% y el 20-30%, respectivamente).
- —
Localización del tumor: en extremidades mejor que en esqueleto axial.
- —
Grado de necrosis tumoral tras la QT preoperatoria .
- —
Volumen tumoral.
- —
Presencia de tumor en los márgenes quirúrgicos.
El sarcoma de Ewing (SEw) se integra en el grupo de TFEw, al que pertenecen el SEw, el tumor neuroectodérmico primitivo (PNET), el SEw atípico y el tumor de Askin (PNET de la región torácica). Son el segundo tumor óseo primario por frecuencia en niños, y no son hereditarios ni se asocian a síndromes malformativos. Se originan en las células posganglionares parasimpáticas pertenecientes al sistema nervioso parasimpático autónomo, y en todos se ha identificado la misma anomalía cromosómica, la t(11;22).
La cirugía continúa siendo la pieza clave del tratamiento, pero a pesar del buen control local de la enfermedad, por sí sola no es capaz de curarlos dada la alta incidencia de micrometástasis en el momento del diagnóstico. La introducción de QT adyuvante con doxorubicina y metotrexato, y posteriormente junto a ifosfamida y cisplatino, consiguió elevar la SLE a los 5 años hasta un 70%, presumiblemente gracias a su capacidad para erradicar las micrometástasis presentes al diagnóstico17. El empleo de ciclos con estos agentes como QT neoadyuvante (preoperatoria), aparte de la eliminación de la enfermedad microscópica, consigue un cierto grado de destrucción tumoral, con la consecuente disminución de volumen de la neoplasia, lo que facilita la cirugía extirpadora y reparadora; asimismo, permite evaluar la respuesta histológica como factor pronóstico16. Varios protocolos incluyen QT de consolidación postoperatoria, aunque su beneficio no está plenamente demostrado2.
La RT desempeña un papel secundario, dada la resistencia del OS a la radiación ionizante; si bien, se emplea en localizaciones axiales en las que la cirugía no es posible, o en casos de OS multifocal.
Lectura rápida
Los TFEw son algo más frecuentes en las extremidades que en el esqueleto axial. El dolor suele ser el síntoma inicial, localizado en la región afectada, y puede haber síntomas generales (fiebre). Casi en un tercio de los casos se produce metástasis al diagnóstico, que se localiza en el pulmón, los huesos y la médula ósea. En la radiografía simple, el hueso afectado presenta un patrón moteado difuso, con predominio de áreas líticas, en el que es típica la imagen en “capas de cebolla”. La TC proporciona información sobre la cortical y los cambios en la estructura ósea, y la RM es la técnica radiológica de elección para valorar la extensión ósea y extraósea del tumor.
Con la enfermedad metastásica, se han de intentar combinaciones agresivas de QT y cirugía, a pesar de lo cual la SLE oscila entre un 16 y un 53%. Los únicos factores pronósticos independientes, según análisis multivariante, han sido el número y la completa resección quirúrgica de las metástasis18. Así, los esquemas actuales de tratamiento se elaboran de la forma siguiente, independientemente de que se trate de enfermedad localizada o diseminada (fig.3):
- 1.
QT neoadyuvante con ifosfamida, metotrexato a altas dosis, cisplatino y doxorubicina.
- 2.
Tratamiento quirúrgico, en el que se debe resecar completamente el tumor en bloque (compartimento óseo y partes blandas circundantes, incluidos los trayectos cutáneos y subcutáneos de las biopsias iniciales), con márgenes de seguridad, procediendo después a la reconstrucción del sistema musculoesquelético mediante injertos óseos (autólogos o alogénicos de cadáver) o prótesis. Son cirugías complejas que requieren que las realice un cirujano ortopeda con experiencia en el tratamiento de tumores óseos pediátricos. En el mismo acto se debe intentar, en caso de que existan, la exéresis quirúrgica de todas las metástasis posibles, pues de ello depende en gran medida la supervivencia en los casos de enfermedad diseminada.
- 3.
Se puede administrar QT de intensificación posquirúrgica con los mismos agentes que en la fase inicial si la respuesta histológica ha sido favorable.

Hasta un 40% de los pacientes con enfermedad localizada recaen en los primeros 3 años tras el diagnóstico, generalmente en el pulmón. Alcanzar la remisión completa nuevamente es el factor pronóstico más importante. La supervivencia global (SG) tras la recaída oscila entre el 13 y el 57%16. La segunda línea de QT no está bien definida, puesto que el OS presenta poca sensibilidad a agentes distintos de los citados. El tratamiento con megadosis de QT, seguido de rescate con autotrasplante de progenitores hematopoyéticos (ATPH), no ha ofrecido resultados esperanzadores19. En el momento actual, se están ensayando nuevos tratamientos, como topotecán, gemcitabina, interleucina 2, interferón alfa, antagonistas de la hormona de crecimiento y tratamiento génico.
Tumores de la familia de EwingEn 1921, James Ewing describió el SEw como “un tumor óseo sensible a la RT y originado en el endotelio vascular”. Costó más de 60 años establecer que su origen histológico real es neural, y actualmente se integra en el grupo de TFEw, a la que pertenecen el SEw, el tumor neuroectodérmico primitivo (PNET), el SEw atípico y el tumor de Askin (PNET de la región torácica). Todos ellos pueden localizarse en cualquier hueso, y también hay formas extraóseas.
EpidemiologíaLos TFEw son el segundo tumor óseo primario por frecuencia en niños, por detrás del OS, y suponen el 5% de las neoplasias infantiles. Es muy raro en la raza negra. Más del 50% de los casos aparecen en la segunda década de la vida, pero entre el año y los 10 años de edad son más frecuentes que el OS20. No son hereditarios ni se asocian a síndromes malformativos.
HistopatogeniaSe originan en las células posganglionares parasimpáticas pertenecientes al sistema nervioso parasimpático autónomo, ampliamente distribuidas en tejidos blandos y óseos, lo que explica sus diversas localizaciones. Los diferentes TFEw se distinguen entre ellos por el grado de diferenciación neural: escasa en el SEw típico, marcada en el PNET e intermedia en el SEw atípico. con técnicas de inmunohistoquímica, se comprueba que expresan marcadores neurales, como la enolasa neuroespecífica y la S-100; generalmente la glucoproteína de superficie MIC-2 también es positiva21.
En todos los TFEw se ha identificado la misma anomalía cromosómica, la t(11;22), o una de sus variantes, detectable en el 90-95% de los casos por reacción en cadena de la polimerasa22,23. Es una translocación entre los cromosomas 11 (gen EWS) y 22 (gen FLI1). Ambos se fusionan en el cromosoma 22 y surge un gen quimérico EWS-FLI1, causante del inicio y el mantenimiento de la tumorogenia al producir desregulación en los genes encargados de la proliferación y la diferenciación celular. Según la localización del punto de rotura del gen FLI1, hay 2 tipos de translocaciones llamadas tipo 1 (T1) y tipo 2 (T2)24.
ClínicaLos TFEw pueden originarse en cualquier hueso, y en tejidos blandos. Son algo más frecuentes en las extremidades que en esqueleto axial (45%). Suele haber afectación de los tejidos blandos adyacentes al tumor. Los síntomas son muy inespecíficos y poco alarmantes, de forma que más de la mitad de los pacientes llevan más de 6 meses de evolución antes de que se realice el diagnóstico. El dolor suele ser el síntoma inicial (96%), localizado en la región afectada, pero puede irradiar si hay afectación radicular; no es continuo y se atribuye fácilmente a algún tipo de traumatismo. La tumefacción aparece de forma más tardía, y suele ser el motivo de la consulta que lleva al diagnóstico del tumor. A veces hay síntomas generales, como fiebre (21%). No es infrecuente que una fractura patológica sea el inicio de la enfermedad25. Otra sintomatología dependerá de la localización del tumor: derrame pleural en los tumores de la pared costal, dolor radicular en tumores vertebrales, o incluso síndrome de compresión medular, por lo que puede producirse una lesión medular permanente si no se actúa a tiempo. Los tumores pélvicos alcanzan un gran tamaño hasta que originan sintomatología por compresión mecánica o nerviosa, como problemas de esfínteres.
Los TFEw tienen una capacidad elevada de metastatizar a distancia: después de diagnosticar casi un tercio de los casos hay una enfermedad diseminada. Las metástasis más frecuentes aparecen en pulmón (38%), seguidas por las óseas (31%) y en médula ósea (11%)26.
LaboratorioLos datos analíticos son totalmente inespecíficos. Puede haber cierto grado de anemia, aumento de VSG, y la LDH se eleva en relación con la masa tumoral existente.
RadiologíaLos hallazgos son muy indicativos de tumor maligno, pero no son patognomónicos. En la radiografía simple, el hueso afectado presenta un patrón moteado difuso, con predominio de áreas líticas. Es típica la imagen en “capas de cebolla”, causada por la existencia de múltiples capas de reacción perióstica con neoformación ósea. Puede haber triángulo de Codman. En huesos planos, el único hallazgo puede ser la esclerosis (fig. 1).
La TC proporciona información sobre la cortical y los cambios en la estructura ósea. La RM es la técnica radiológica de elección para valorar la extensión ósea y extraósea del tumor27. En la fase T1, se aprecia la afectación de la médula del hueso y la posible existencia de focos lejanos al tumor primario (lesiones skip), y en la fase T2, la invasión cortical y de tejidos blandos (fig. 2).
Lectura rápida
El diagnóstico es histológico. El estudio de extensión tumoral requiere realizar una gammagrafía ósea con tecnecio 99 y con talio 201, una TC de pulmón y 2 biopsias de médula ósea.
PronósticoLa supervivencia global se sitúa en torno al 60-75%. Se asocian a un mejor pronóstico: la localización del tumor en extremidades o huesos prescindibles, la ausencia de enfermedad metastásica y el grado de necrosis del tejido tumoral tras la quimioterapia inicial superior al 90%.
Lectura rápida
Para el tratamiento, debe seguirse un protocolo específico para los TFEw, multidisciplinario, en el que se combinen quimioterapia sistémica con las medidas locales (radioterapia y/o cirugía), y hay que estratificar a los pacientes en grupos de riesgo, según la localización del tumor y la presencia de metástasis, e intensificar el tratamiento en los grupos de más riesgo.
La PET permite evaluar la actividad metabólica del tumor, pero no supera a la RM en cuanto a datos sobre la extensión tumoral local.
DiagnósticoEs un diagnóstico histológico; la biopsia, al igual que en el OS, es preferible llevarla a cabo con trócar dentro de la zona que después se resecará, preferiblemente por el mismo cirujano que realizará después la intervención quirúrgica para resecar el tumor, y con el máximo cuidado para evitar contaminación de los tejidos circundantes por células neoplásicas. La punción aspirativa con aguja fina generalmente aporta poco material para el estudio, aunque puede ser suficiente para valorar metástasis dudosas.
Estudio de extensión tumoralLa gammagrafía ósea con 99Tc es una técnica muy sensible para definir la extensión del tumor primario y descubrir metástasis óseas (tumores multicéntricos). Con talio 201 la captación es más intensa que con el 99Tc, y además tiene más sensibilidad que éste para valorar los cambios producidos en el tumor tras el tratamiento.
Con la TC de pulmón se detectan las metástasis pulmonares, y el estudio de extensión se completa con biopsias de médula ósea en 2 lugares alejados del tumor primario para determinar si hay metástasis a ese nivel.
Diagnóstico diferencialSe hará con otros tumores malignos (OS, fibrohistiocitoma maligno, metástasis óseas, etc.) y con tumores benignos (tabla 2).
PronósticoAunque en las últimas décadas el pronóstico de los TFEw ha ido mejorando gracias a los tratamientos más agresivos, y a un mejor control local del tumor, la SG se sitúa en torno al 60-75%28–30.
Se asocian a un mejor pronóstico, y por tanto una supervivencia mayor:
- —
Edad inferior a 15 años31,32.
- —
Tumores localizados en extremidades o huesos prescindibles frente a los situados en esqueleto axial y pelvis (el 69% frente al 44%)33.
- —
- —
Fusión EWS-FLI1 de tipo 1.
- —
Enfermedad localizada: la presencia de metástasis al diagnóstico disminuye considerablemente las posibilidades de curación (SLE, 25-30%)31,32,34. El pronóstico es mejor para los pacientes con afectación pulmonar exclusivamente (SLE, 29-34%) que para los que presentan metástasis en médula ósea y/o huesos (14-28%)31,32,35.
- —
Grado de necrosis del tejido tumoral tras la QT inicial superior al 90%36,37.
Los pacientes deben incluirse en protocolos específicos para los TFEw, en los que se combinan la QT sistémica con las medidas locales (RT y/o cirugía). Por ello, es imprescindible un tratamiento multidisciplinario, y contar con la cooperación entre oncólogos, cirujanos y radioterapeutas infantiles con experiencia en el tratamiento de tumores óseos malignos infantiles. Los pacientes se estratifican en grupos de riesgo según los factores pronósticos al diagnóstico (localización del tumor y presencia de metástasis), y se intensifica el tratamiento en los grupos de más riesgo.
La QT sistémica es imprescindible para curar un TFEw; antes de su empleo como adyuvante a la RT o la cirugía, sólo el 10% de los pacientes sobrevivían 5 años, pues los TFEw son siempre enfermedades diseminadas desde el comienzo, y no basta con la resección del tumor, por radical que sea (amputación) para controlar la enfermedad. Los agentes más activos contra los TFEw son ciclofosfamida, ifosfamida, doxorubicina, actinomicina y etopósido, y la mayoría de los protocolos actuales se basan en combinaciones de estos 5 citostáticos38.
El esquema de tratamiento (fig. 3) es similar al del OS:
| Malignización | Observaciones |
|---|---|
| 1% de adultos | Tumor óseo benigno más frecuente en pediatría |
| Sí en asociaciones sindrómicas (Ollier y Maffucci) | |
| No | |
| No | Forma de histiocitosis de células de Langerhans con afectación esquelética exclusiva |
| No | Lesión recurrente hasta en el 30% de los casos |
| No | Regresión espontánea |
| No | Asociación a pubertad precoz y pigmentación cutánea en el síndrome de Albright |
- 1.
QT inicial neoadyuvante para reducir el volumen tumoral y eliminar las micrometástasis.
- 2.
Tratamiento local: es imprescindible, pues sólo con la QT no se consigue curar un TFEw. Puede realizarse la cirugía39, como en el OS, o se puede optar por la RT, pues estos tumores son radiosensibles y con ella se puede eliminar completamente el tumor32,40,41. La elección de RT o cirugía para el tratamiento local depende de la localización del tumor, su tamaño y de la edad del niño. En general, los huesos “prescindibles” (peroné, clavícula, costillas, etc.) se resecan sin más. Los tumores localizados en las extremidades suelen ser extirpables, y mediante prótesis e injertos (autoinjertos o aloinjertos de cadáver) se conserva la extremidad con un buen grado de funcionalidad. Si el hueso no es resecable, o la cirugía supone una grave mutilación o deformidad estética, se utiliza la RT como tratamiento local.
- 3.
Tras el tratamiento local, se administra más QT para eliminar tumor residual. También se puede administrar RT como complemento a la cirugía si el grado de necrosis tumoral tras la QT inicial es inferior al 90%, o hay células tumorales en los márgenes de la resección.
Para los tumores de mal pronóstico, tras el tratamiento local se puede administrar una QT mieloablativa (megaterapia) con ATPH, puesto que los TFEw responden muy bien a los agentes alquilantes que se usan en el acondicionamiento (busulfano y melfalán)42.
En cuanto a las metástasis pulmonares, si no desaparecen con el tratamiento QT, se puede administrar RT pulmonar35.
RecaídasOcurren sobre todo durante los primeros 2 años desde el diagnóstico, pero aun a los 6 años de evolución, las curvas de supervivencia no han alcanzado la meseta31,43. Son más frecuentes las recaídas metastásicas, en las que el pronóstico es malo, con supervivencias del 20-25%, y es peor cuanto más temprana sea (SLE, 4-5% si la recidiva ocurre dentro de los primeros 2 años del diagnóstico, y casi 0 si se produce durante el tratamiento). En todos los casos se debe administrar QT, y no limitarse a actuar sobre el tumor44. La combinación de topotecán-ciclofosfamida produce respuesta aproximadamente en el 35% de los pacientes, y es un tratamiento que está actualmente en investigación45.
Lectura rápida
La quimioterapia inicial neoadyuvante reduce el volumen tumoral y elimina las micrometástasis; el tratamiento local puede hacerse con cirugía y/o radioterapia, ya que son tumores radiosensibles, y tras éste se administra más quimioterapia para eliminar tumor residual. El trasplante autólogo de progenitores hematopoyéticos se realiza en los tumores de mal pronóstico. Los efectos secundarios del tratamiento a corto y medio plazo son consecuencia de la quimioterapia: pancitopenia, mucositis, toxicidad neurológica, hepática, renal, etc. A largo plazo, la radioterapia ocasiona un mal desarrollo del hueso en crecimiento (deformidades, asimetrías), y las secuelas tras la cirugía se ocasionan por la disparidad de crecimiento con la extremidad contralateral, o por problemas con los injertos o prótesis (falta de unión, fractura o reabsorción del injerto, e infecciones). También hay un riesgo mayor de desarrollar segundas neoplasias, como leucemias, osteosarcomas, fibrosarcomas, etc.

Los efectos inmediatos del tratamiento son consecuencia de la QT, e incluyen pancitopenia grado III-IV, y mucositis en grado variable; el metotrexato puede causar toxicidad neurológica y hepática. A medio plazo, la administración de ifosfamida puede originar una tubulopatía renal y el cisplatino puede producir pérdida auditiva.
Las secuelas por la RT pueden ser graves, pues el hueso en período de crecimiento que recibe RT se desarrolla mal, debido a pérdida de células proliferantes en la placa de crecimiento y de células que sintetizan la matriz ósea. En general, las regiones óseas que han recibido RT antes de que el niño haya finalizado su crecimiento no crecerán al mismo ritmo que el resto del esqueleto, de forma que, según la localización de la lesión, se producirán deformidades y asimetrías más o menos graves.
Las secuelas tras la cirugía se ocasionan por la disparidad de crecimiento con la extremidad contralateral, falta de unión, fractura o reabsorción del injerto, e infecciones. Si se ha amputado la extremidad inferior, hay prótesis que permiten al niño mantener una actividad física adecuada con pequeño impacto social y emocional, pues proporcionan una buena calidad de vida. Para la extremidad superior, hay prótesis mioeléctricas que permiten una buena función.
Como ocurre con otras neoplasias infantiles, en los supervivientes de un tumor óseo hay una incidencia mayor de cáncer que en la población normal. Como segundas neoplasias, se han descrito leucemias no linfoblásticas y síndromes mielodisplásicos, osteosarcomas, fibrosarcomas, fibrohistiocitomas, y otros varios tumores del adulto, generalmente desarrollados en regiones previamente radiadas, con un período de latencia medio de 7,6 años desde el diagnóstico del tumor inicial32,46.
Por todo ello, es preciso realizar un seguimiento prolongado de todos los pacientes que sobreviven a un tumor óseo maligno47.